摘要
从简单构建单元区域选择性获得缺电子和高取代吡咯仍然具有挑战性。本文结果表明,O-乙烯基羟胺能够通过氮杂-Michael/氧氮杂-Cope[3,3]-σ迁移重排/芳构化级联反应与活化炔烃实现吡咯的从头构建。这一单锅转化在室温下使用催化量温和有机碱快速进行,生成二氢吡咯半缩胺中间体,可进一步直接转化为吡咯或有选择性地功能化。该方法适用于一系列O-乙烯基羟胺和活化炔烃偶联试剂,能够以前需要苛刻条件或危险乙炔基方法才能获得的取代模式。这些结果确立了O-乙烯基羟胺作为合成高度功能化吡咯的多功能砌块。
氮杂环是现代治疗剂中最普遍的结构基序之一,吡咯单元在生物活性天然产物、药物、功能材料和农用化学品中广泛出现。[1]吡咯亚结构嵌入关键生物分子中,如维生素B12、叶绿素、血红素和胆汁色素,突显了它们在生物功能和分子识别中的核心作用(1A)。[2]这一广泛的重要性推动了超过一个世纪以来旨在从开链砌块构建吡咯核的方法开发。[3]然而,从简单前体区域选择性获得高取代和缺电子吡咯仍然具有挑战性。特别是,许多已建立的方法依赖于预官能化底物,在温和且操作简单的条件下对取代模式控制有限。这些局限性凸显了对新策略的需求,即能够从readily available的砌块从头构建高度功能化的吡咯骨架。
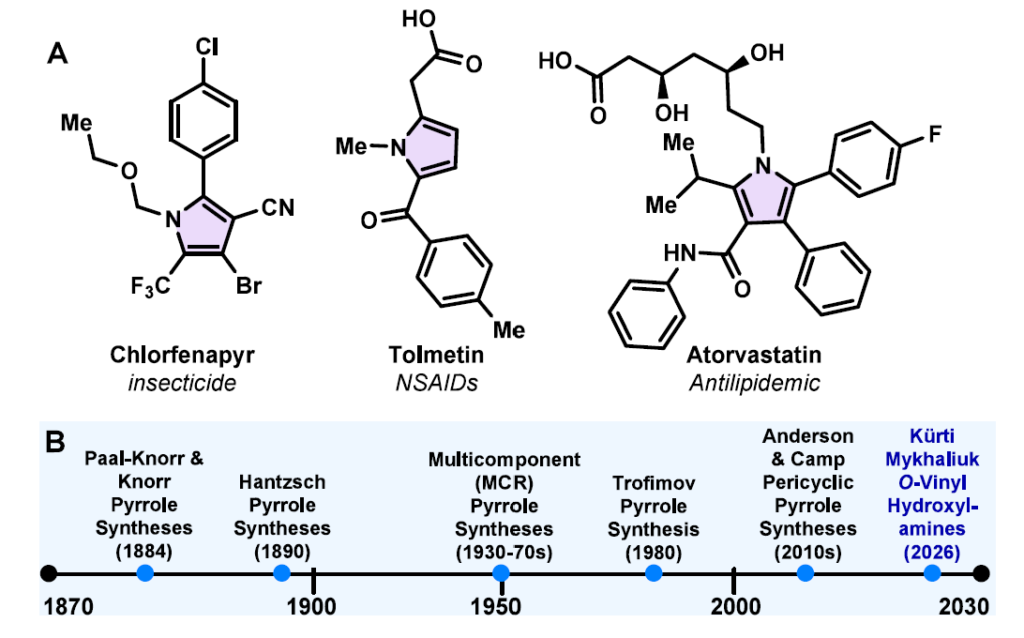
从开链前体合成吡咯已开发出大量策略(图1B和方案1)。[4]经典方法,包括Paal−Knorr(方案1A),[5]Knorr(方案1B),[6]Hantzsch(方案1C),[7]van Leusen(方案1D)[8]和Barton-Zard(方案1F)[9]反应,为取代吡咯的合成提供了可靠途径,但通常依赖于预官能化底物,如1,4-二羰基化合物,[10]α-氨基酮或活化异氰化物。最近的多组分和环合方法扩大了可获得的取代模式范围;[11]然而,这些方法通常需要剧烈条件,特别是对于缺电子和稠密取代吡咯。
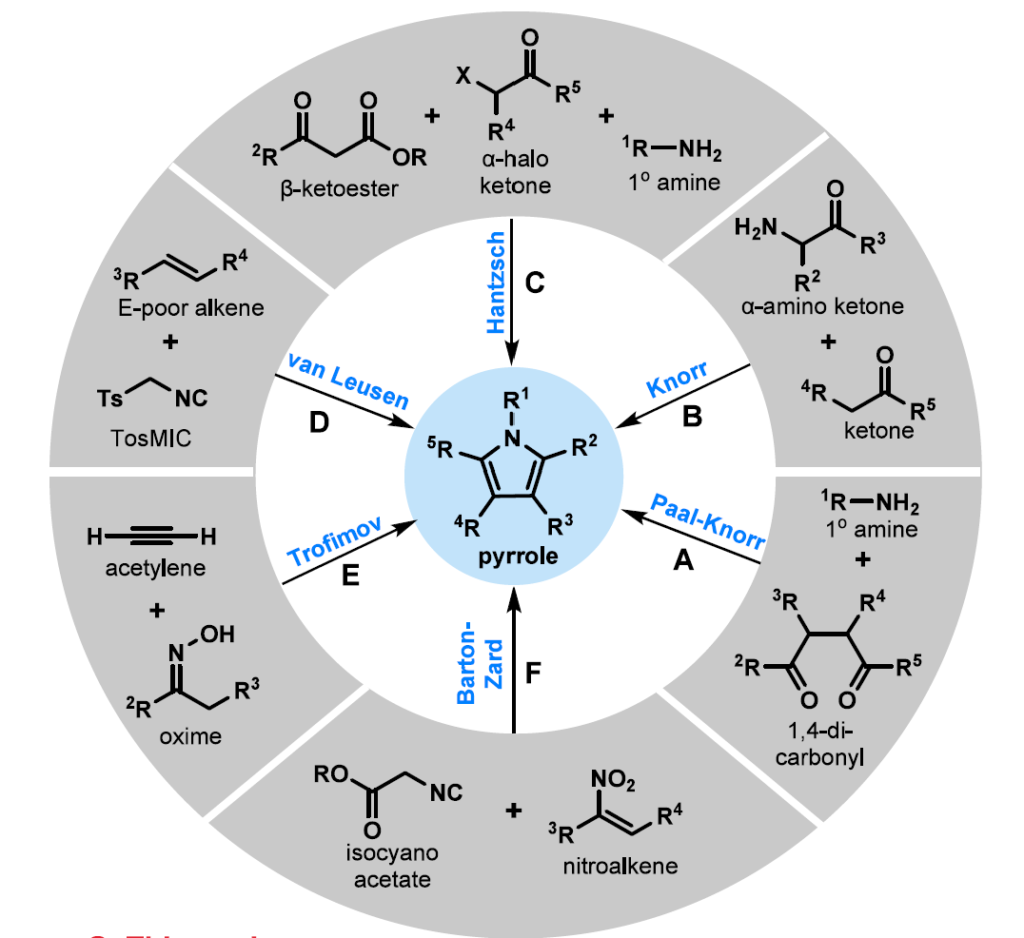
基于σ迁移重排的策略已作为概念上独特的吡咯构建方法崭露头角。[12]在此背景下,Trofimov证明酮肟与乙炔的偶联可以通过O-乙烯基肟的连续互变异构/氧氮杂-Cope重排生成吡咯,确立了这一途径的可行性(方案1E)。[12b]然而,对乙炔气体、强碱性条件和高温的要求显著限制了该方法的实际范围和官能团耐受性。
尽管取得了这些进展,但从简单且易于获得的砌块在温和条件下合成高度功能化、缺电子吡咯的区域选择性和模块化策略仍然匮乏。最近,我们报道了单N-保护的O-乙烯基羟胺试剂作为氮杂环合成的多功能"两碳/一氮"环加成试剂。[13]这些试剂通过N-芳基化引发的级联过程实现了7-氮杂吲哚啉和7-氮杂吲哚的高效获得,该过程经由N-芳基-O-乙烯基羟胺中间体的[3,3]-σ迁移重排进行。基于此类反应性,我们设想O-乙烯基羟胺的N-乙烯基化可以通过类似的重排途径实现吡咯的从头构建。
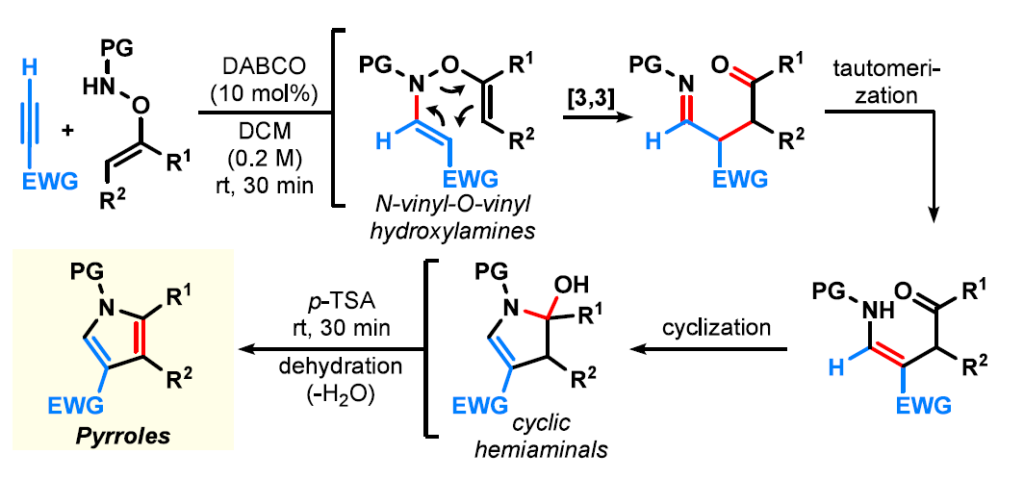
在此,我们报道了从单N-保护的O-乙烯基羟胺(方案1G)合成缺电子吡咯的一锅法合成。该转化通过氮杂-Michael加成到活化炔烃上生成N-乙烯基-O-乙烯基羟胺,随后经历氧氮杂-Cope重排,然后环化和脱水以提供吡咯产物。值得注意的是,两步成键过程在催化量温和碱的存在下室温快速进行。与之前需要乙炔气体和/或苛刻条件的方法相比,该方法为吡咯提供了直接获得途径,包括其他方法难以获得的取代模式。这些结果确立了氮杂-Michael/氧氮杂-Cope重排名为吡咯合成的快速且操作简单的途径,并进一步强调了O-乙烯基羟胺作为杂环构建多功能砌块的潜力。综上所述,这种转化反映了 O-乙烯基羟胺的不同反应性范式,即这些试剂作为模块化环加成试剂而非单一杂环支架的前体。
受氮杂-Michael加成化学先例的启发,[14]其中氮亲核性腙在温和碱催化条件下与活化炔烃反应,[15]我们评估了单N-保护的O-乙烯基羟胺是否可以参与类似过程。活化炔烃因其易于获得且与无过渡金属条件相容而成为理想的偶联试剂。
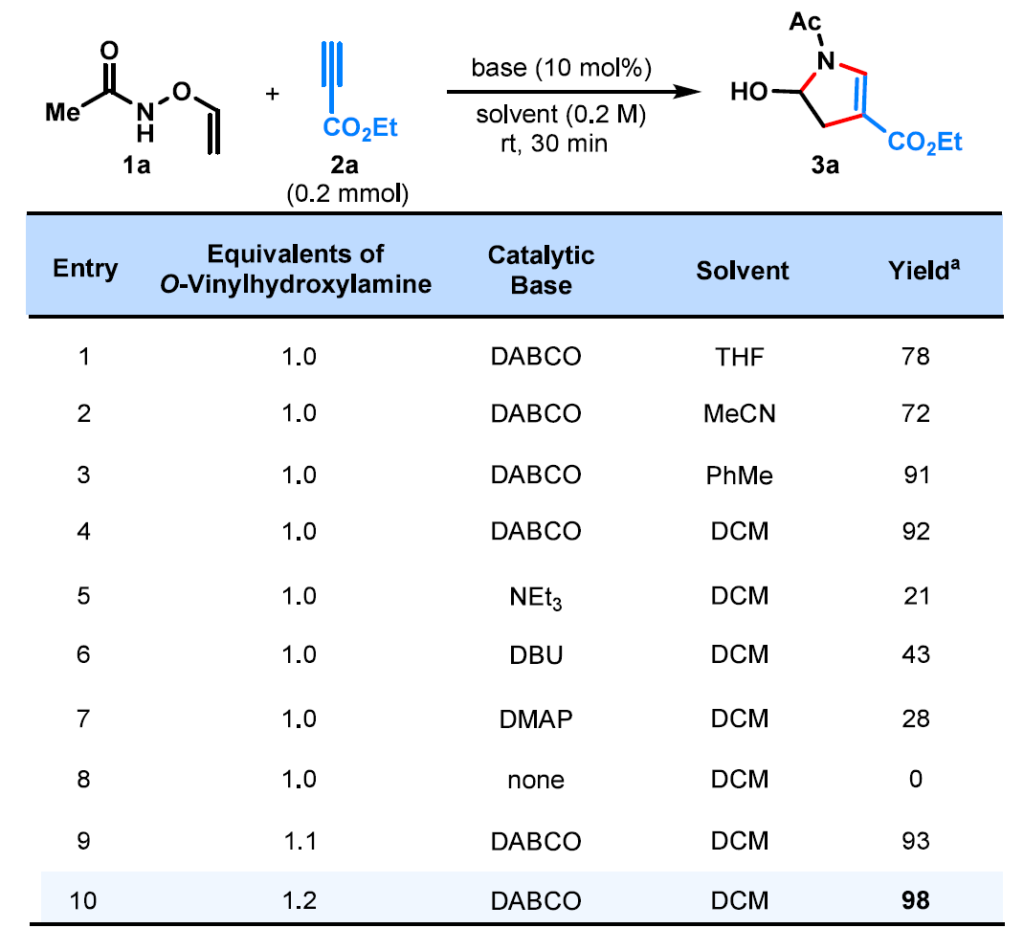
在THF中,10 mol% DABCO存在下,将O-乙烯基羟胺(1a)与丙炔酸乙酯(2a)反应,以78%的产率得到相应的4,5-二氢吡咯3a(表1,第1项)。溶剂效应评估表明,使用DCM可将产率提高至92%(第4项)。替代有机碱(三乙胺、DBU、DMAP)效果欠佳,且在没有碱的情况下未发生反应(第5−8项)。将O-乙烯基羟胺的投料比增至1.2当量进一步将产率提高至98%(第10项)。
二氢吡咯半缩胺中间体3a既可以分离出来,也可以在加入p-TSA后在一锅法中易于实现芳构化,在1小时内提供相应的吡咯。在建立了优化的反应条件后,我们考察了该转化的适用范围,其适用于一系列官能团,并以中等至优异的产率获得吡咯。
在这些条件下,单N-保护的O-乙烯基羟胺与末端炔烃选择性反应,而二取代炔烃无反应性。值得注意的是,当使用未保护的母体O-乙烯基羟胺时,可以参与二取代活化炔烃,这凸显了反应性特征依赖于N-取代方式。
我们首先考察了N-保护基的影响。在评估的各种保护基中,商业可得的N-乙酰基衍生物(1a)表现最佳,以90%的产率提供目标吡咯(方案2,4a),而N-Boc和N-Cbz衍生物也可行,尽管效率有所降低(4b,4c)。
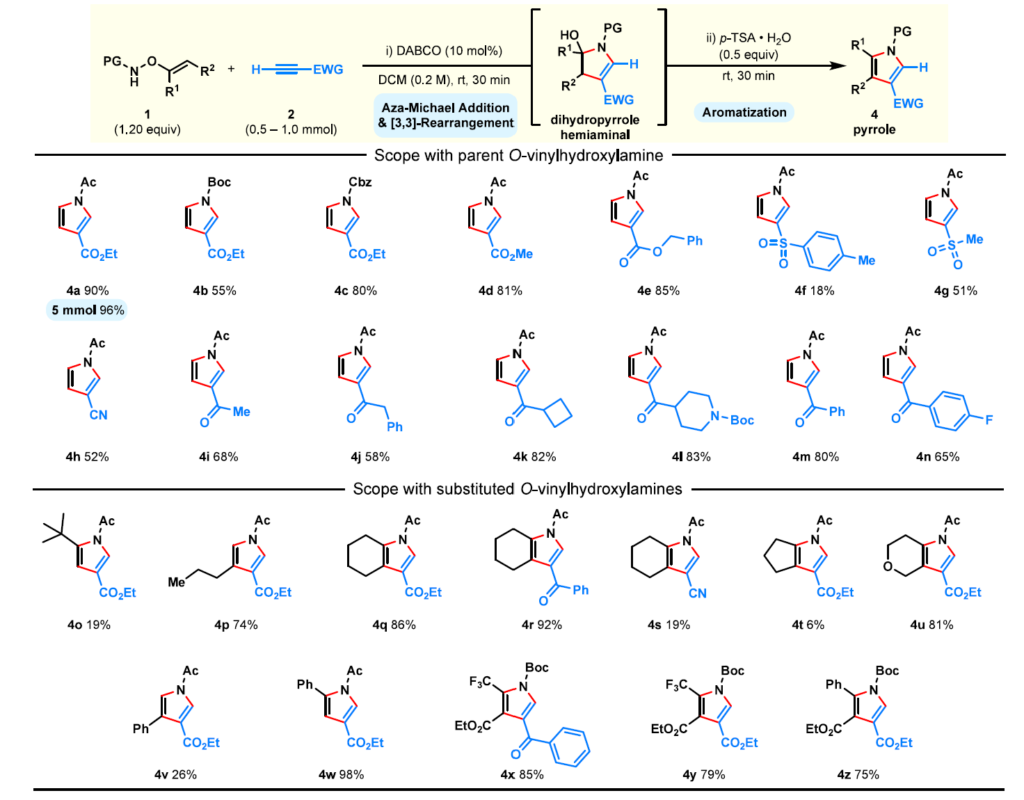
随后使用N-乙酰基O-乙烯基羟胺考察了炔烃成分的范围。一系列缺电子末端炔烃易于参与反应,以中等至优异产率提供C-3取代吡咯(4a,4d−4n)。丙炔酸酯效果尤为突出(4a,4d),且该反应易于放大,在5 mmol规模上以96%产率获得4a。苄基丙炔酸酯的反应结果同样良好(4e)。
接下来考察了具有其他吸电子基团的炔烃。磺酰基取代炔烃的反应效率较低(4f,4g),而氰基取代炔烃仍是可行的偶联伙伴(4h)。酮基取代炔烃,包括无环和环状变体,均表现出良好的兼容性(4i−4l),芳基酮衍生物(4m,4n)亦是如此,表明该方法对含多种羰基官能团具有良好的耐受性。
接下来我们探索取代的O-乙烯基羟胺以合成更多功能化的吡咯(4o−4z)。[13,16]这些试剂允许在C-2、C-3和C-4位进行系统性的变化,提供二取代和三取代产物。烷基取代羟胺以可变产率提供相应的吡咯(4o,4p),而环状底物则形成稠合吡咯骨架(4q−4u),包括带有不同吸电子取代基的实例。芳基取代的O-乙烯基羟胺(4v,4w),以区域异构和立体化学纯的烯基前体制备,提供了不同的吡咯产物,确立取代基在乙烯基片段上的位置决定了区域化学结果。
通过氧杂-Michael加成从活化炔烃制得的N-Boc-O-乙烯基羟胺也是有效的偶联试剂,使获得带有CF3和酯基等强吸电子取代基的吡咯成为可能(4x−4z)。
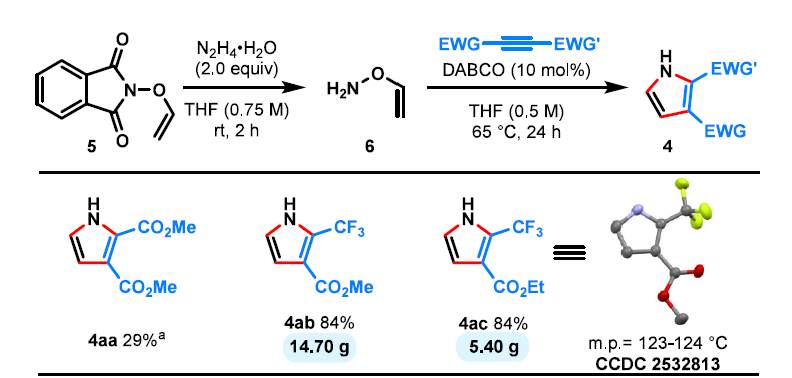
最后,带有两个吸电子基团的二取代活化炔烃也可以使用未保护的母体O-乙烯基羟胺参与反应(方案3,6)。尽管该试剂易挥发,但可以方便地以稀溶液形式操作并高效促进吡咯的形成,包括克级规模的合成(方案3,4ab,4ac)。
综上所述,这些结果确立了一个模块化平台,兼容广泛范围的炔烃和O-乙烯基羟胺试剂,实现了结构多样吡咯的系统组装。除这一范围外,观察到的反应性突显了O-乙烯基羟胺作为可编程环加成试剂而非仅限于单一转化类型的底物的重要作用。
为了进一步扩展这一平台的合成实用性,我们考察了替代的芳构化条件以及相应中间体的后续功能化。作为p-TSA脱水条件的替代方案,我们开发了一种使用三乙胺和甲磺酰氯将二氢吡咯半缩胺中间体转化为相应吡咯的方法(4ad;方案4A)。这种单锅法氮杂-Michael加成/芳构化序列在5 mmol规模上进行了展示,以81%产率获得4ad。
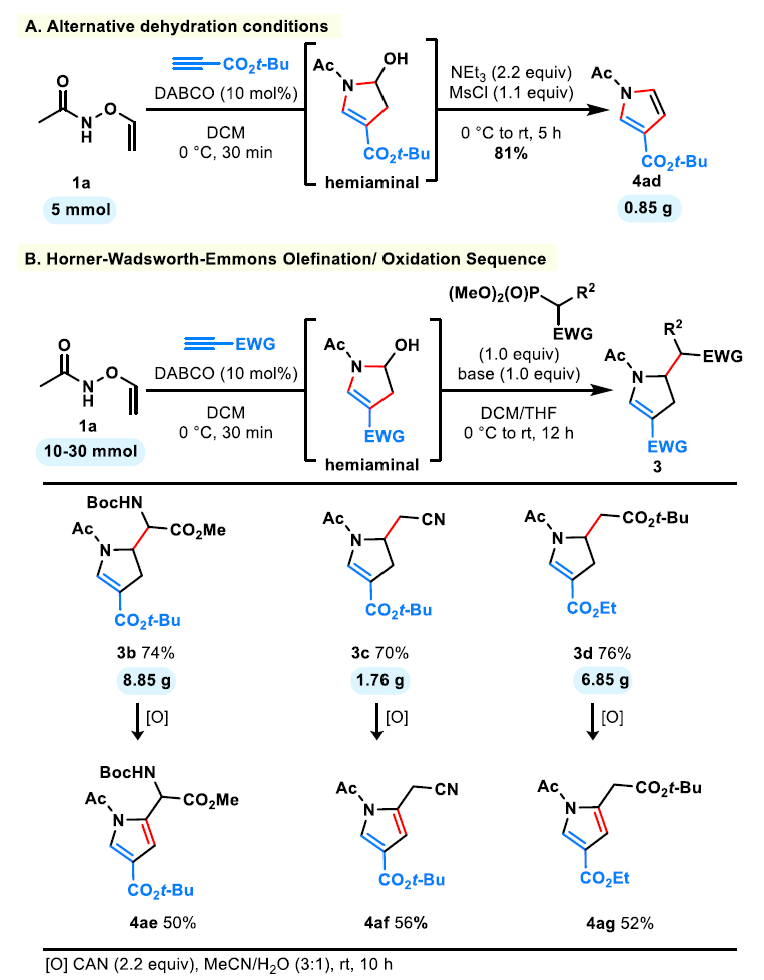
利用二氢吡咯半缩胺中间体(因N-保护基而稳定),我们探索了其合成实用性(方案4B)。这些中间体与其相应的醛存在平衡,这与已知的半缩胺类结构的性质一致,由此实现了HWE烯基化反应/氮杂-Michael加成序列在C-5位形成新的C−C键,得到烷基化二氢吡咯(3b−3d)。随后用硝酸铈铵(CAN)氧化(脱氢)提供相应的吡咯(4ae−4ag)。
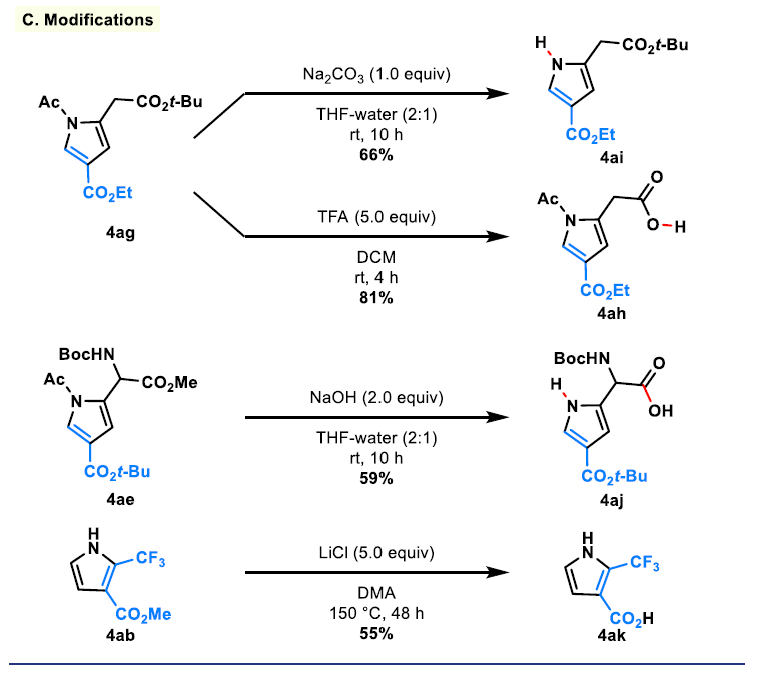
在碱性条件(Na2CO3,THF/H2O)下去除N-乙酰基保护基,提供N−H吡咯4ai,产率66%(方案4C)。其他衍生化反应进一步展示了产物的合成灵活性。甲基酯4ae的碱介导水解得到4aj,而4ag中叔丁酯的选择性酸介导裂解得到羧酸4ah,未影响乙基酯或N-乙酰基。4ab与LiCl在高温下处理实现皂化反应,得到4ak。值得注意的是,CF3取代的吡咯在酸性和碱性条件下均表现出显著的敏感性。这种不稳定性可能源于CF3取代体系通过醌甲烷型中间体发生水解的文献报道倾向,特别是当位于额外吸电子取代基附近时。[17]在当前体系中,C-2位CF3基和C-3位酯基(4ab)的组合使吡咯对该类转化高度活化,导致在典型条件下迅速分解。尽管存在这种内在不稳定性,该底物仍可耐受Krapcho型条件,[18]实现了相应羧酸的形成,突显了这一选择性功能化的实用窗口。
总之,我们开发了一种快速且实用的合成方法,通过 O-乙烯基羟胺与活化炔烃之间的氮杂-Michael/氧氮杂-Cope重排/芳构化级联反应合成缺电子吡咯。该转化在温和、无过渡金属条件下室温下进行,为单、二和三取代吡咯提供了直接获得途径。该方法易于放大规模,能够以传统方法难以获得的取代模式进行合成,包括带有相邻未取代位置的吡咯。此外,所得的二氢吡咯和吡咯中间体易于进一步多样化,彰显了这一平台的合成实用性。综上所述,这些结果确立了单N-保护的O-乙烯基羟胺作为多功能环加成试剂,并定义了一个反应性图谱,实现了高度功能化吡咯骨架的模块化获得。