1、摘要
本文开发了一种以市售N-叔丁氧羰基保护的内酰胺和内酯为起始原料,合成(氧杂)氮杂螺[2.n] 烷砌块的高效可拓展方法。以Tebbe烯化反应结合环丙烷化反应为关键步骤,成功构建了包含五至七元杂环的螺环核心结构,且该方法的可拓展性已在62克制备规模中得到验证。后续通过非对映体分离和简便的官能团转化,得到了一系列衍生物,包括羧酸、胺类以及偕二氟取代类似物,从而拓展了对这类研究较少的大环骨架的获取途径。实验脂溶性测定结果表明,偕二氟环丙烷环的螺并环化修饰可显著提高化合物的log P,其作用效果与三氟甲基(CF₃)基团相当。单晶X射线衍射研究和出口向量图(EVP)分析完成了代表性化合物的结构表征,证实了其三维空间结构,并为其在电子等排体替换中的潜在应用提供了依据。综上,本研究为螺环砌块的克级制备和理化性质分析建立了可靠的研究平台,这类螺环结构在药物化学领域具有重要的应用价值。
2、引言
螺环片段凭借其能为先导化合物引入构象刚性和三维结构的特性,在现代药物研发中备受关注。与平面芳香族骨架不同,螺环结构可打破分子的平面性,减少分子间堆积作用,并为官能团的引入提供非共面的出口向量。这些特性通常能改善化合物的理化性质,包括提高溶解度和优化脂溶性分布,进而提升药物的吸收、分布、代谢和排泄(ADME)特性。螺环中心还能调节分子的构象柔性,使化合物能更精准地与蛋白质结合口袋相互作用。
在螺环化合物这一大类中,含环丙烷环的螺环母核尤为引人注目。环丙烷单元空间位阻小——仅略大于偕二甲基基团,却具有高度的环张力,赋予化合物独特的电子效应和构象效应。从理化性质角度来看,环丙烷取代基可降低晶体堆积效率、熔点,并提高水溶性。例如,螺环丙基糖模拟物已被开发为稳定、不可水解的生化工具,可用作抑制剂或底物类似物,适用于共结晶研究。
氮杂螺烷是螺环化合物中的一类优势骨架,常作为六元杂环的饱和电子等排体,其药物应用价值已通过多个实例得到印证。5-氮杂螺[2.4]庚烷衍生物被报道为有效的食欲素受体拮抗剂和多巴胺D3受体拮抗剂,同时也存在于已获批的丙型肝炎药物雷迪帕韦和抗生素西他沙星中,其中螺环核心在降低药物毒副作用、提高药物溶解度方面发挥关键作用。更大的6-氮杂螺[2.5]辛烷骨架则被开发为胰高血糖素样肽-1(GLP-1)受体激动剂、M4毒蕈碱型乙酰胆碱受体拮抗剂和CB2受体抑制剂。含环丙烷的螺环片段还可作为甲基的替代基团,用于制备选择性L-岩藻糖苷酶抑制剂、调节哌啶环的构象,或作为具有抗骨肉瘤活性的人乳酸脱氢酶5抑制剂的结构单元。
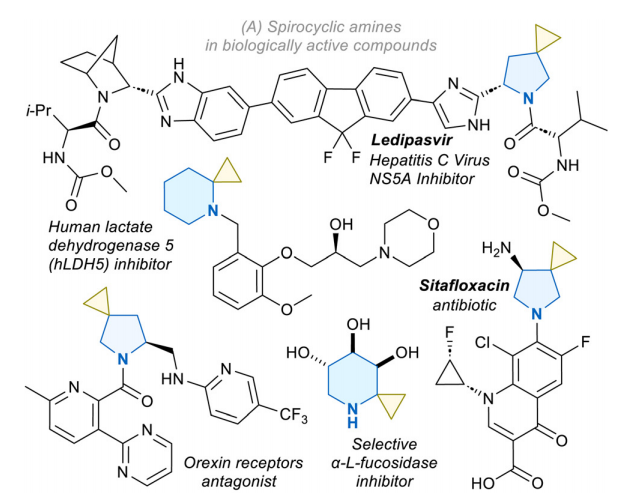
尽管这类化合物的应用价值已得到证实,但氮原子直接与螺环连接位点相邻的氮杂螺[2.n]烷(氮近位型),相较于其研究更为深入的异构体,仍未得到充分探索,目前仅在研究阶段的药物中发现其存在。造成这一现象的原因可能与该类骨架的合成可行性有关。现有氮杂螺烷的合成策略包括内酰胺的Kulinkovich反应及其与腈的中断型修饰反应、环丙胺的分子内环化反应,也有部分烯烃官能团化的合成路线被报道,但这些方法对N-叔丁氧羰基保护的底物往往效果不佳。
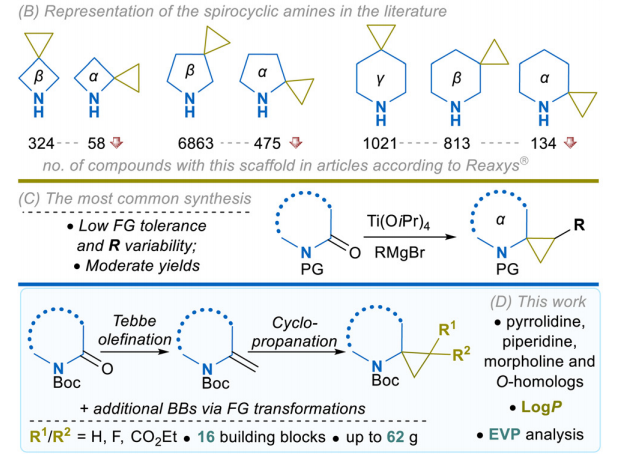
本课题组近期报道了一种可拓展的4-氮杂螺[2.3]己烷衍生物合成方法,以Tebbe烯化反应为起始,后续经铜催化的环丙烷化或偕二氟环丙烷化反应得到目标产物。本研究将该方法进一步拓展,合成了一系列(氧杂)氮杂螺[2.n]烷化合物,目标为大环中含5~7个重原子的饱和杂环。该合成路线以环丙烷化反应为核心转化步骤,所得模块化砌块可进行进一步的结构修饰。本策略为研究较少的氮近位型螺环胺类化合物提供了高效的合成途径,为其构效关系研究和在药物化学中的广泛应用奠定了基础。
3、结果与讨论
合成研究
本研究的合成工作以市售N-叔丁氧羰基吡咯烷酮(1a)与按已有方法制备的Petasis试剂(2)的烯化反应为起始。研究发现,使用1.3倍当量的Petasis试剂储备液,可使起始原料内酰胺1a完全转化,得到目标环外烯胺衍生物3a。将有机溶剂小心蒸干后,粗产物用正己烷研洗,蒸干有机萃取液后得到纯度约80%的烯胺3a;该方法在190克1a的制备规模下仍能保持反应效率。由于3a本身具有不稳定性,尝试通过蒸馏或色谱法进一步纯化均未成功,因此所得产物直接用于下一步反应。不过,制备得到的烯胺3a及其高同系物在-10℃下可储存1个多月;储存1年后,仅吗啉衍生物及其高同系物基本保持稳定,而吡咯烷和哌啶衍生的烯烃3a和3b则发生分解。
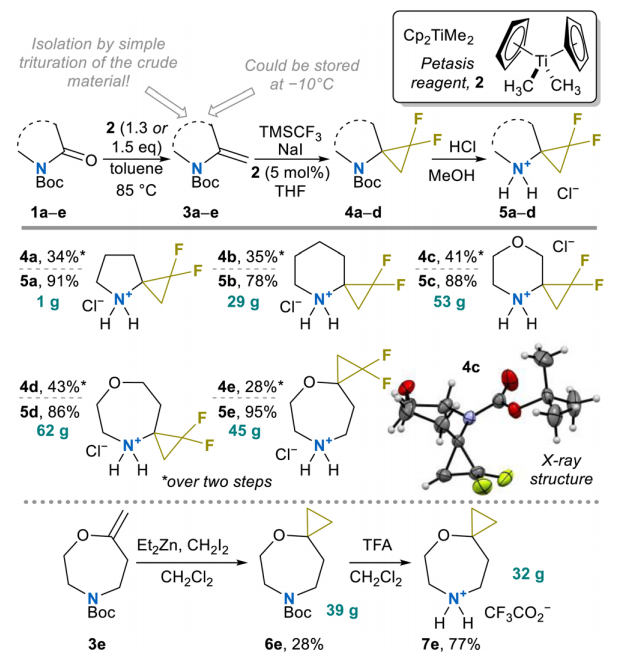
通过本课题组自主开发的“缓慢滴加”实验方案,将化合物3a与Ruppert–Prakash试剂(TMSCF₃)反应,成功引入偕二氟环丙烷片段。经柱色谱纯化后,在50克制备规模下,目标偕二氟取代螺环化合物4a的分离收率为49%。最后,通过酸性条件脱除N-叔丁氧羰基保护基,几乎定量得到胺类化合物5a的盐酸盐,该产物可通过乙醚简单研洗实现纯化。
随后,将该合成序列拓展至高同系物和含氧内酰胺,包括哌啶酮(1b)、吗啉酮(1c)和1,4-氧氮杂卓酮(1d)衍生物。在所有实验中,经正己烷研洗后,均可得到收率和纯度均理想的相应烯胺3b~3d(氢核磁共振谱显示纯度约80%,制备规模可达140克),但为使起始原料完全转化,所需Petasis试剂2的用量有所增加(相对于3a的130摩尔%,此处需150摩尔%)。后续经偕二氟环丙烷化和叔丁氧羰基脱保护反应,得到目标螺环胺类化合物5b~5d,均以克级规模制备(单次实验产量29~62克),总收率理想。为进一步探索该方法的适用范围,本研究以酯类化合物1e为底物进行实验。烯化反应高效进行,得到相应的乙烯基醚3e(纯度约80%,制备规模125克);经二氟环丙烷化和叔丁氧羰基脱保护后,在45克制备规模下以良好的总收率得到胺类化合物5e的盐酸盐。另一条合成路线中,3e在Simmons–Smith反应条件下与二乙基锌-碘甲烷(Et₂Zn-CH₂I₂)反应,得到螺环醚6e,但收率一般;6e经酸性脱保护后得到三氟乙酸盐7e,制备规模为32克。
此外,烯烃3a与重氮乙酸乙酯在铜催化下发生环丙烷化反应。实验发现,以乙酰丙酮铜(Cu (acac)₂,5摩尔%)为催化剂、1.5倍当量重氮酯为原料时,反应几乎仅生成反式异构体8a(收率42%,仅含微量顺式异构体)。将该反应条件应用于烯烃3b和3c时,反应效果不佳;而以苯络合三氟甲磺酸铜([CuOTf]₂・C₆H₆)为催化剂时,可在150克制备规模下得到相应的酯类产物,非对映体比例分别为2:1(8b)和1:1(8c)。尽管反应的非对映选择性较低,但可通过快速柱色谱法分离异构体混合物,以理想收率得到顺式8b、8c和反式8b、8c的单一非对映体(收率包含分离过程中回收的原料)。
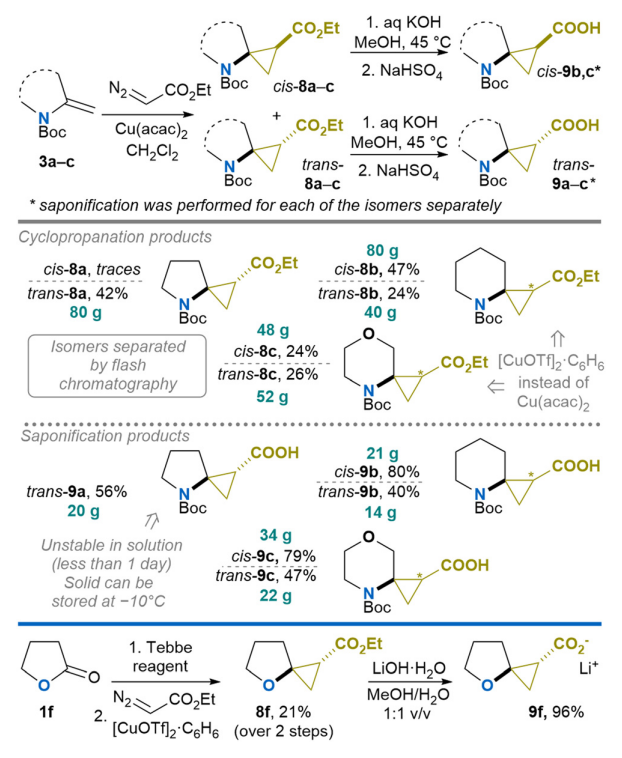
将合成得到的所有酯类产物(顺式8b、8c和反式8a~8c)在常规碱性水解条件下(甲醇中的2摩尔/升KOH水溶液)反应。经酸化、萃取和重结晶后,得到目标N-叔丁氧羰基保护的氨基酸(顺式9b、9c和反式9a~9c),收率40%~80%,制备规模可达34克。所幸水解过程中未发生异构体化。反式9a~9c的收率低于顺式9b、9c(分别为40%~56%和79%~80%),这一现象可归因于供体-受体型环丙烷在碱性条件下易发生开环反应的固有特性。化合物的稳定性表征进一步证实了这一点:反式9a在氘代氯仿溶液中,1小时内发生部分分解,24小时内完全降解;且该化合物在固态室温下储存时也会发生明显分解,但在-10℃下可实现长期稳定储存(超过1年)。
酯基还可用于制备含氧螺环衍生物。以γ-丁内酯(1f)为底物,经烯化反应得到烯烃3f(因具有挥发性,以甲苯溶液形式使用),后续经环丙烷化反应得到酯8f,收率21%。遗憾的是,上述水解方案对该化合物无效;而使用化学计量的水合氢氧化锂,可成功得到目标产物9f的锂盐,且该产物具有良好的储存稳定性(室温固态储存1个月无分解迹象)。
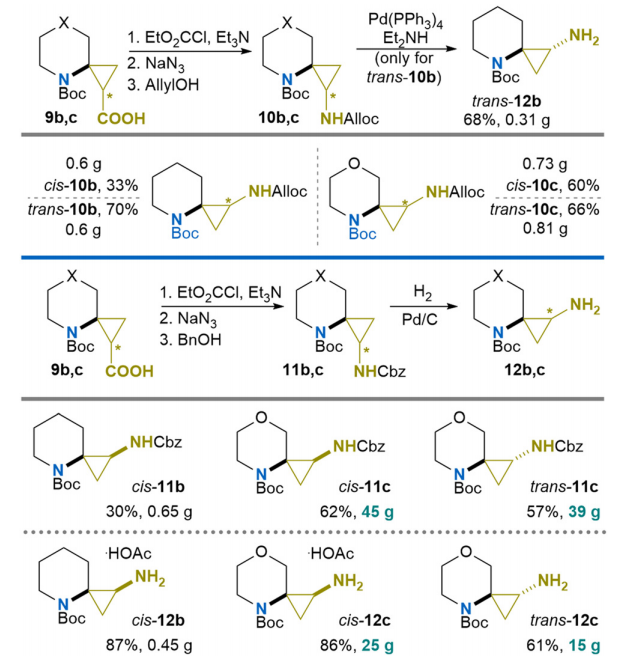
得到一系列羧酸产物后,本研究进一步探索了其经Curtius重排反应转化为N-叔丁氧羰基单保护二胺的方法。最初,选择烯丙氧羰基(Alloc)作为新生成伯氨基的正交保护基。经典Curtius重排条件下生成的异氰酸酯中间体与烯丙醇淬灭反应,得到相应的氨基甲酸酯。对于稳定性最差的底物反式9a,该方法无法得到目标产物,仅生成复杂的开环产物混合物;而羧酸9b和9c(包括顺式和反式异构体)的副反应较少,可在45克制备规模下以33%~70%的收率得到相应的氨基甲酸酯10b和10c。但后续的烯丙氧羰基脱保护步骤效率较低:反式10b与四三苯基膦钯-二乙胺反应,仅以68%的收率得到产物反式12b;其他底物(顺式10b、顺式10c和反式10c)均无法得到目标二胺衍生物。其中,顺式12b和顺式12c在硅胶纯化过程中不稳定;反式12c虽性质稳定,但无法通过柱色谱法与杂质分离。
因此,本研究调整策略,在Curtius重排步骤中通过生成苄氧羰基(Cbz)衍生物,完成了二胺衍生物顺式12b、12c和反式12c的合成。以苄醇作为异氰酸酯中间体的淬灭试剂,在45克制备规模下以30%~62%的收率得到N-苄氧羰基氨基甲酸酯顺式11b、顺式11c和反式11c;经苄氧羰基还原脱除后,以中至高收率得到目标产物,制备规模可达25克。对于顺式12b和顺式12c,在氢化反应前加入1当量乙酸可提高收率和纯度,最终产物以乙酸盐形式分离得到。
脂溶性研究
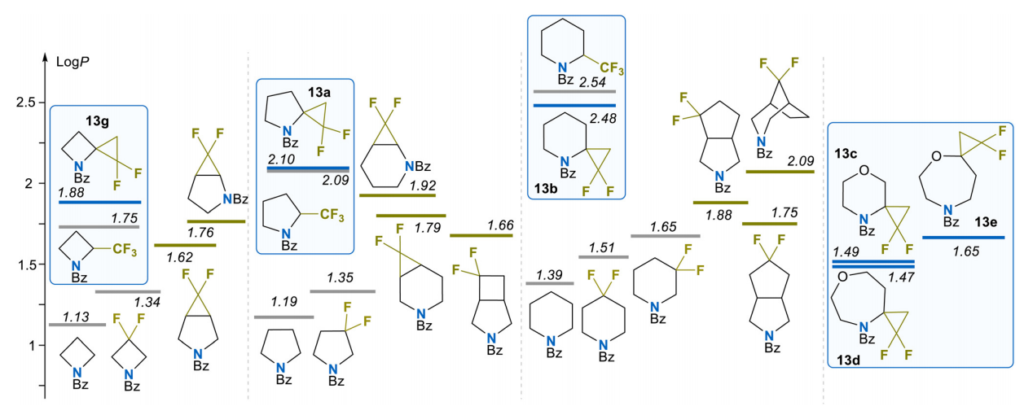
采用经典的“摇瓶法”结合高效液相色谱分析,测定了苯甲酰胺类化合物13a~13e的分配系数对数值(log P),上述苯甲酰胺由相应的盐酸盐5a~5e经标准酰胺合成方法制备。为进行对比,实验同时测定了化合物13g的log P值,并引入了已有报道的单环、稠环或桥环双环类似物的相关数据。
研究发现,偕二氟环丙烷单元的螺并环化修饰,可使氮杂环丁烷、吡咯烷和哌啶衍生物的log P值分别提高0.75、0.91和1.09个单位。该提升效果远大于单纯偕二氟取代的作用(log P值提升0.12~0.26个单位),且与α-三氟甲基基团的作用效果高度接近(log P值提升0.62~1.15个单位)。因此,在本研究的化合物系列中,螺并环化的偕二氟环丙烷可作为三氟甲基取代基的刚性电子等排体。此外,化合物13g、13a和13b的脂溶性高于其所有含相同碳原子数的稠环或桥环双环异构体(包括稠合偕二氟环丙烷衍生物)。这一结果表明,氟化螺环单元对化合物脂溶性具有显著影响,这一现象可能源于该刚性片段降低了分子的溶剂可接触性。
正如预期,氧原子的引入可抵消上述脂溶性的提升效应。值得注意的是,与已有报道的单环类似物相比,该抵消作用稍弱(本研究中log P值降低约1.0个单位,而单环化合物中为1.5~1.7个单位)。此外,异构体13d和13e的脂溶性存在细微差异,log P值相差0.16个单位。
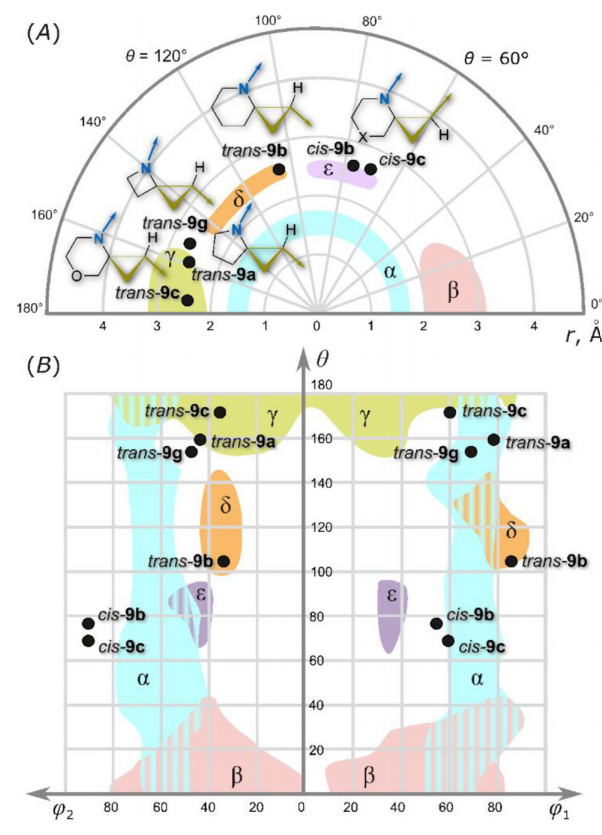
出口向量分析
基于N-叔丁氧羰基保护的氨基酸(顺式9b、9c和反式9a~9c)以及偕二氟取代胺类衍生物(4a、4c、4d)的单晶X射线衍射数据,结合已有报道的反式9g和5g的相关数据,本研究通过出口向量图(EVP)分析完成了标题螺环骨架的结构表征。
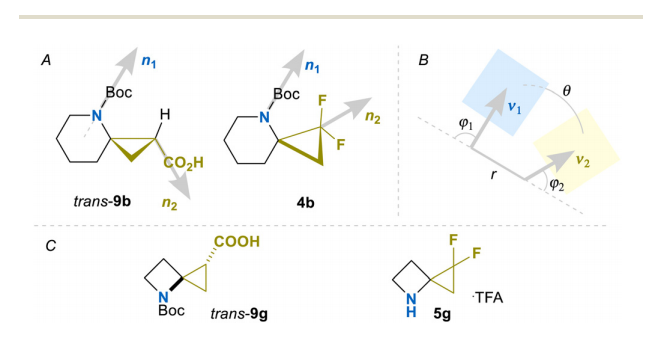
出口向量图分析的原理是利用出口向量模拟连接在目标骨架上的官能团:对于氮原子和二氟亚甲基(CF₂)片段,分别以碳-氮-碳键角和氟-碳-氟键角的角平分线定义出口向量方向;对于羧基(COOH),则直接以碳-碳键作为出口向量。生成的向量n₁和n₂的相对空间分布可通过四个几何参数明确描述:向量起点间的距离r、平面角φ₁和φ₂、二面角θ。
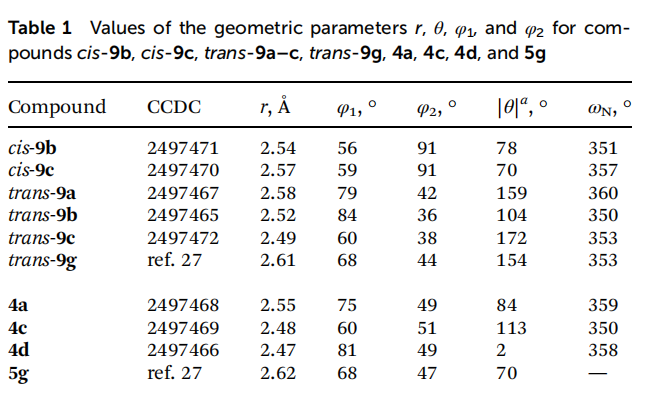
研究发现,参数r的取值范围为2.47~2.62埃,环张力越大的体系(即环尺寸越小、且为顺式异构体),r值越大。描述与氮原子相连的出口向量n₁取向的平面角φ₁变化范围较小,为56°~84°;而与官能团或偕二氟片段相连的向量n₂对应的平面角φ₂,在顺式9系列化合物中为91°,在反式9系列中为36°~42°,在4系列和5g中为47°~51°。二面角θ的变化范围最大:顺式9系列为70°~78°,反式9系列为104°~172°,4系列和5g为2°~113°。显然,φ₂和θ的数值差异与环丙烷环上官能团的两种可能取代位置相关。
值得特别关注的是4-氮杂螺[2.5]辛烷衍生物反式9b与其7-氧杂类似物反式9c的参数差异。对二者构象的深入分析发现,反式9b中环丙烷环上连接羧基的C-1原子处于平伏键位置;而在反式9c中,该基团则处于直立键位置。顺式异构体中未观察到该现象,其构象高度相似。
将9系列化合物的相关数据绘制为出口向量图,可直观体现该类骨架在电子等排体替换中的应用潜力。顺式1,4-二取代4-氮杂螺[2.5]辛烷骨架(顺式9b)及其7-氧杂类似物(顺式9c)位于ε区域附近,该区域为1,3-二取代吡咯烷和哌啶的特征区域,但二者的平面角φ₁/φ₂存在显著偏差。反式1,4-二取代4-氮杂螺[2.3]己烷、4-氮杂螺[2.4]庚烷和7-氧杂-4-氮杂螺[2.5]辛烷(分别对应反式9g、反式9a和反式9c)位于γ区域附近,该区域常见于1,3-二取代氮杂环丁烷、1,4-二取代哌啶、反式1,3-二取代环丁烷和反式1,4-二取代环己烷;同时,这类化合物的φ₁参数存在一定偏差,且二面角θ值更低,表明该类螺环衍生物具有更高的三维结构特征。最后,反式1,4-二取代4-氮杂螺[2.5]辛烷骨架(反式9b)位于δ区域,该区域为反式1,3-二取代环戊烷和环己烷的特征区域。
本研究以氮原子为中心的价键角之和(ω_N)表征N-叔丁氧羰基保护衍生物中氮原子的锥化程度。结果显示,ωN的取值为350°~360°,表明该类化合物中的氮原子几乎无锥化现象。
4、结论
本研究提出了一种以易得的内酰胺或内酯为起始原料,克级合成(氧杂)氮杂螺[2.n]烷砌块的简便方法。该合成路线以Petasis烯化反应和环丙烷化反应为关键步骤,可直接克级制备多种螺环骨架(分离产物规模可达62克)。通过进一步的结构修饰,得到了一系列具有应用价值的砌块,包括保护氨基酸、二胺以及偕二氟取代胺类化合物,其中涵盖了目前研究较少的大环类似物。
对模型衍生物的脂溶性测定表明,偕二氟环丙烷环的螺并环化修饰可显著提高化合物的log P值,其提升效果远高于其他稠合或桥环双环偕二氟环丙烷体系,且与三氟甲基取代基的作用效果相似,提示螺并环化的偕二氟环丙烷可作为三氟甲基基团的刚性电子等排体。
基于X射线衍射实验结构的出口向量图(EVP)分析表明,标题螺环骨架可作为1,3-和1,4-二取代哌啶或环己烷(以及药物研发中较少使用的其他饱和环)的扭曲型三维替代物。
综上,本研究为这类尚未得到充分探索的(氟化)螺环母核提供了实用的合成路线,深入揭示了氟化修饰和环尺寸对其结构和理化性质的影响,凸显了其作为新一代药物化学砌块的应用潜力。
本文中涉及的(氧杂)氮杂螺[2.n]烷砌块与相关衍生物,Enamine均有现货供应。