导读:这篇2025年bioRxiv预印本是中美乌合作的结构药物研发论文:用6500万分子超大对接发现TAAR1激动剂,经冷冻电镜验证,最终得到强效、无僵直副作用的精神分裂症候选药,由UCSF、上海药物所、Enamine、杜克大学四家单位主导完成。
1、摘要
精神分裂症是一种严重的精神疾病,当前治疗药物主要靶向多巴胺受体和5-羟色胺受体。这类药物常引发副作用,且在不同患者中的疗效差异显著。痕量胺相关受体1(TAAR1)可调控单胺能信号传导,已成为极具潜力的替代药物靶点。为发现新型TAAR1配体,我们针对TAAR1激活态构象对6500万个小分子进行计算机虚拟对接,并实验验证了55个排名靠前的化合物。结果筛选出14个具有TAAR1激动活性的分子,其效价介于中纳摩尔级至微摩尔级,且均为激动剂。这种高度的功能选择性,可能源于激活态TAAR1正构位点呈现的紧凑构象。尽管该构象利于优先筛选激动剂,但模拟研究表明,若过度优化初始筛选命中率,可能会牺牲后续的亲和力优化空间。本研究通过先导化合物优化,获得了纳摩尔级效力的激动剂,其对接预测的结合模式经冷冻电镜实验验证。其中3个激动剂具有优异的脑暴露量,效力显著优于在研药物乌洛塔罗特,足以开展行为学研究。这3个化合物均能有效逆转小鼠中安非他明诱导的前脉冲抑制缺失(精神分裂症模型),且不会引发传统抗精神病药常见的僵直副作用。
2、引言
痕量胺相关受体1(TAAR1)是一种G蛋白偶联受体(GPCR),近年来因其在调控单胺能神经传递中的作用而备受关注¹。TAAR1可被β-苯乙胺(β-PEA)、酪胺、色胺等痕量胺类,以及安非他明、甲基苯丙胺(METH)等多种合成精神兴奋剂激活²⁻³,其内源性神经递质尚未明确。与主要表达于嗅觉区域的其他TAAR家族成员不同⁴,TAAR1广泛分布于大脑,尤其在腹侧被盖区(VTA)、中缝背核(DRN)等单胺能核团中高表达⁵⁻⁶。这种分布与其对多巴胺能⁵和5-羟色胺能⁷系统(包括奖赏回路与情绪调控)的调节作用高度吻合⁸。这些特性使TAAR1成为精神分裂症、焦虑、抑郁、精神兴奋剂成瘾等精神疾病的潜在治疗靶点⁶,⁹⁻¹¹。事实上,兼具强效TAAR1激动活性与5-HT₁A受体活性的药物SEP-363856(乌洛塔罗特),已被开发为精神分裂症新型治疗药物¹¹⁻¹²。乌洛塔罗特旨在同时改善精神分裂症的阳性与阴性症状,同时降低传统抗精神病药常见的副作用¹³⁻¹⁴。尽管早期研究结果喜人,但该药物在Ⅲ期临床试验中未达到主要终点,研发进程终止。
尽管乌洛塔罗特研发受挫,TAAR1仍是治疗精神分裂症的重要靶点¹⁵⁻¹⁹,目前正通过基于受体同源模型的大规模分子对接筛选,探索全新化学骨架的配体¹⁷,²⁰。近期,大规模分子对接技术在多个药物靶点上展现出巨大潜力⁹,²¹⁻³⁴。本研究利用最新解析的TAAR1激活态实验结构,开展新型激动剂的发现工作。通过大规模分子对接,我们获得了效力介于23nM至20μM的TAAR1激动剂,总体筛选命中率(活性化合物数/测试化合物数)达25%(14/55)。我们对其中3个激动剂进行活性与药代动力学优化,最终获得TAAR1效力为1~48nM的优化产物。令人振奋的是,这些化合物同时对5-HT₁A受体具有活性——该受体与TAAR1协同作用可实现抗精神病疗效³⁵⁻³⁶。尽管其对5-HT₁A受体的效力相对较低(260~600nM),但显著优于乌洛塔罗特,且结合分子在脑中达到的微摩尔级游离浓度,该活性具有重要临床意义。凭借高效力与良好的药代动力学特性,我们进一步在小鼠模型中评估了这3个化合物的抗精神病样药效。
3、结果
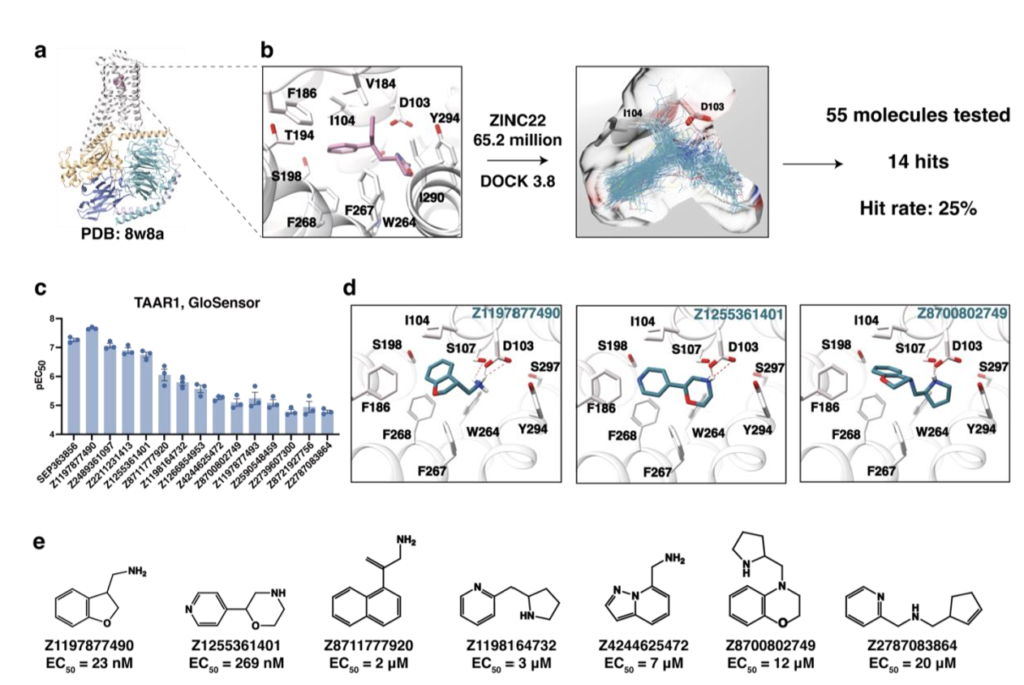
大规模分子对接筛选发现TAAR1激动剂
本研究以TAAR1正构结合口袋为对接靶点,该口袋的构象主要参照激动剂RO5256390在该位点的结合模式(PDB:8W8A)(图1a、b)。采用DOCK3.7软件²¹,³⁸,以预设的热点(“匹配球”)为导向进行分子取向采样。针对每个取向,基于预计算的低能构象系综,最多采样200个配体构象³⁸⁻³⁹。通过泊松-玻尔兹曼法QNIFFT计算的预生成静电势网格,评估配体构型的静电互补性⁴⁰;利用AMBER力场的预生成范德华项,评估非极性互补性⁴¹。同时采用SOLVMAP计算配体的环境依赖性去溶剂化能⁴²。
与纯实验研究类似,在开展前瞻性对接筛选前,对照计算是重要的合理性验证手段⁴³。我们通过测试能否在大量性质匹配³⁸与极值诱饵分子⁴³⁻⁴⁵中,将已知TAAR1激动剂排名靠前,验证采样方法与能量势图的可靠性。通过调整静电计算中低介电与高介电区域的边界,优化静电能与去溶剂化能参数,最终实现对数加权AUC值达31.7(扩展数据图1b)。该优异结果表明,我们的方法能有效区分TAAR1真配体与诱饵分子。
完成对照验证后,我们从ZINC22化合物库⁴⁶中筛选约6500万个类片段阳离子分子(非氢原子数11~19个)进行对接。该计算耗时39993核小时,在1000核集群上运行不到两天即可完成。为获取全新结构化合物,我们从得分前382767的分子中,剔除拓扑结构与已知TAAR1激动剂相似的化合物(ECFP4指纹塔尼莫托系数>0.35的分子均被移除)。为确保配体阳离子与受体正构结合口袋内的D103³・³²、Y294⁷・⁴³、F267⁶・⁵¹、F268⁶・⁵²、I104³・³³、F186ECL2等识别位点直接相互作用,我们采用LUNA软件进行基于相互作用指纹的过滤。该步骤确保阳离子不仅能与D103³・³²区域的受体静电势良好互补(DOCK3.8的打分依据),还能形成可识别的离子对。剩余95126个化合物按拓扑结构聚类(见方法),并通过Chimera软件⁴⁷对排名靠前簇中的最优得分分子进行可视化评估。从中挑选2462个结构多样的化合物(任意两个分子ECFP4塔尼莫托系数≤0.25),评估其结合几何构型与相互作用合理性,最终优先选定66个化合物,其中55个由Enamine公司成功合成,合成满足率达83%(图1b)。
为评估化合物对TAAR1的激活能力,我们首先采用活细胞GloSensor功能实验⁴⁸,在10μM浓度下对55个对接命中化合物进行初筛,检测其通过招募Gs蛋白诱导cAMP产生从而激活TAAR1的能力(图1a)。结果显示,55个化合物中有14个表现出激动活性,其最大效应(Emax)超过乌洛塔罗特(SEP-363856)的30%(图1c、扩展数据图1a、表1)。浓度响应实验证实,这些化合物均为TAAR1激动剂,其半最大效应浓度负对数值(pEC₅₀)为4.7~7.6,最大效应(Emax)为乌洛塔罗特的44%~108%(图1c、扩展数据图1c)。基于效力与新颖性(ECFP4塔尼莫托系数分别为0.27、0.18、0.20),我们优先选择Z1197877490、Z1255361401、Z8700802749三个化合物进行基于结构的优化(图1c~e、扩展数据图2~3、表1~2)。
仅筛选到激动剂
理论上,针对受体激活态或失活态进行对接,应分别倾向于发现激动剂或拮抗剂。尽管我们在部分靶点上观察到该规律²⁵,⁴⁹⁻⁵¹,但多数情况下会同时筛选到激动剂与拮抗剂²¹,²⁸,⁵²⁻⁵⁴。这一结果符合普遍认知:对接主要筛选结合能力,难以区分配体的激活/抑制功能。然而,本研究针对TAAR1激活态对接获得的14个活性化合物均为激动剂,55个测试化合物中无拮抗剂活性分子(补充图1)。为探究该现象,我们对比了28个激动剂(14个新激动剂+14个已知激动剂)与7个已知拮抗剂⁷,⁵⁶⁻⁵⁷,分别对接至TAAR1实验激活态结构与Boltz-2构建的失活态模型⁵⁵时的得分与排名。在TAAR1激活态结构中,28个激动剂中有23个排名进入对接库前0.5%,DOCK得分为-54~-33kcal/mol;而7个拮抗剂中仅1个进入该范围,得分为-45kcal/mol。对接至失活态模型时,激动剂得分变差(负值减小),为-44~-26kcal/mol,平均降低8.5kcal/mol,排名大幅下降;拮抗剂表现则显著提升,最优拮抗剂得分达-46kcal/mol,另有两个拮抗剂得分-42~-35kcal/mol,进入排名前列(扩展数据图4)。
TAAR1激活态对接能优先筛选激动剂、失活态结构相对优先筛选拮抗剂的独特能力,可能源于激动剂与拮抗剂的尺寸差异,以及受体激活/失活态的构象体积差异。TAAR1激动剂通常小于拮抗剂,且受体激活时正构位点会收缩(对接所用的TAAR1模型结构中,该位点收缩更为显著,下文将详述)。α2A肾上腺素受体也存在类似现象:针对其激活态对接同样仅筛选到激动剂²⁵。这表明,当受体激活态与失活态的物理性质(如结合口袋体积)差异显著时,对接预测配体功能(激动剂/拮抗剂)的能力最强;若差异较细微,对接则难以区分配体功能类型。
初始命中化合物优化
为提升初始对接活性化合物的效力,我们采用两种优化策略:一是通过Enamine REAL数据库(如SmallWorld程序,https://sw.docking.org/)搜索结构类似物;二是在合成可行的前提下,对初始命中化合物进行保守修饰,该策略往往效果显著⁴²。
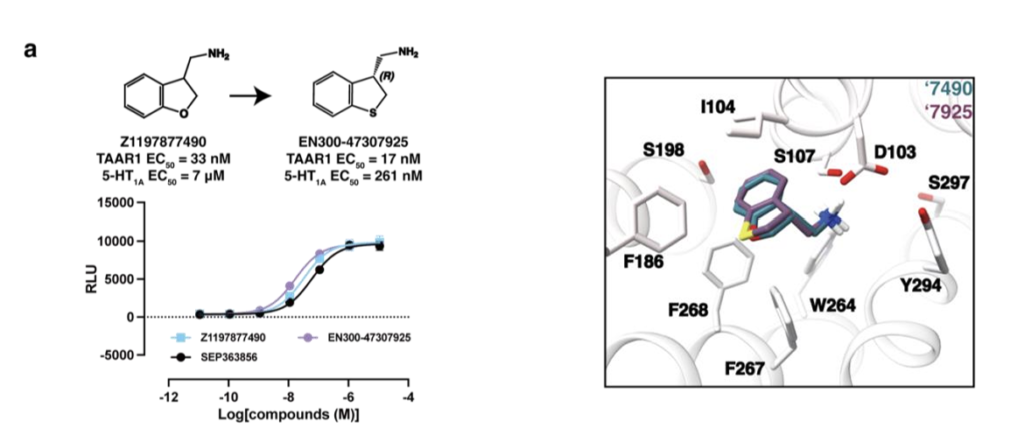
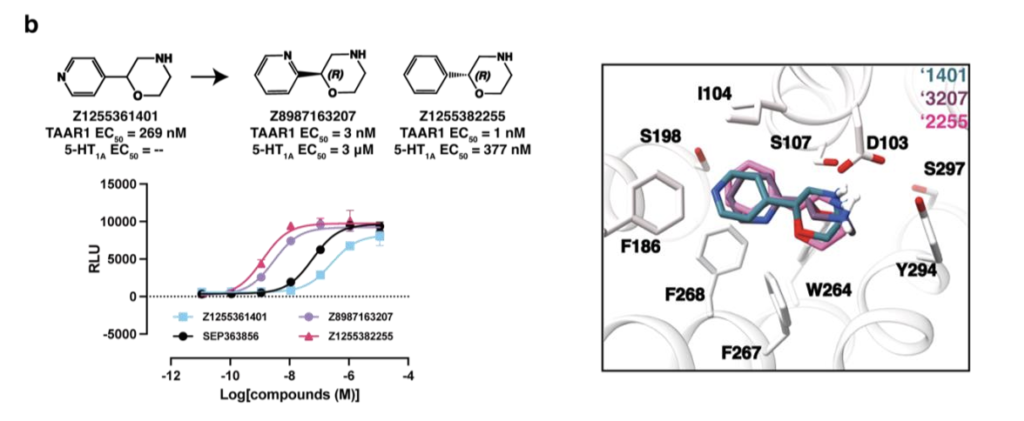
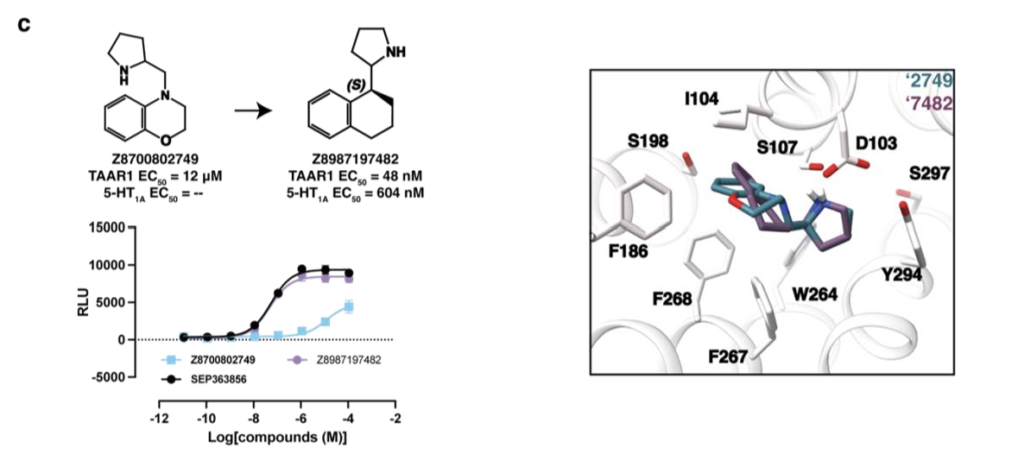
我们从SmallWorld数据库中为Z1197877490筛选2012个类似物,并设计27个衍生小分子;为Z1255361401筛选1707个类似物,设计13个衍生小分子;为Z8700802749筛选873个结构类似物。将所有类似物对接至TAAR1,基于结合效果(得分与可视化评估)优先选择少量化合物进行合成,最终Enamine成功合成5个Z1197877490类似物、6个Z1255361401类似物、1个Z8700802749类似物。母体激动剂Z1197877490的TAAR1 EC₅₀为23nM,将其氧原子替换为碳原子或硫原子等修饰仅小幅提升效力(图2a);立体化学纯化得到EN300-47307925(图2a、扩展数据图2a),对TAAR1效力影响甚微,但使5-HT₁A EC₅₀提升26倍至261nM,接近乌洛塔罗特的10 倍。相反,对EC₅₀为269 nM的Z1255361401进行小幅修饰,可显著提升效力(图2b、扩展数据图2b):将吡啶环内氮原子移位或替换为苯环,活性提升59~107倍;手性拆分获得Z898713207(EC₅₀ 3nM)与Z1255382255(EC₅₀ 1nM),后者对5-HT₁A的EC₅₀为380nM(图2b、扩展数据图2b)。最后,Z8700802749系列需进行较大修饰才能提升效力:将阳离子氮原子向远离苯环的方向移动两个碳原子,活性提升37倍(图2c、扩展数据图3a);手性拆分获得Z8987197482,其TAAR1 EC₅₀为48nM,5-HT₁A EC₅₀为604nM(图2c、扩展数据图3a)。对接构象显示,Z8987197482的连接链缩短一个碳原子,使四氢化萘基团更靠近结合口袋中心,可能增强π-π堆积作用(扩展数据图2c)。
冷冻电镜结构解析
为阐明配体识别的分子机制并为后续优化提供模板,我们解析了TAAR1与两个低纳摩尔级激动剂Z1255382255、Z8987197482的复合物结构(图3a~b、扩展数据图5~6、补充数据表1)。两个复合物的冷冻电镜密度图全局标称分辨率分别为3.14Å与3.12Å。复合物整体结构与已报道的TAAR1实验结构相似,与PDB 8W8A结构的均方根偏差(RMSD)分别为0.492Å与0.846ų⁷。
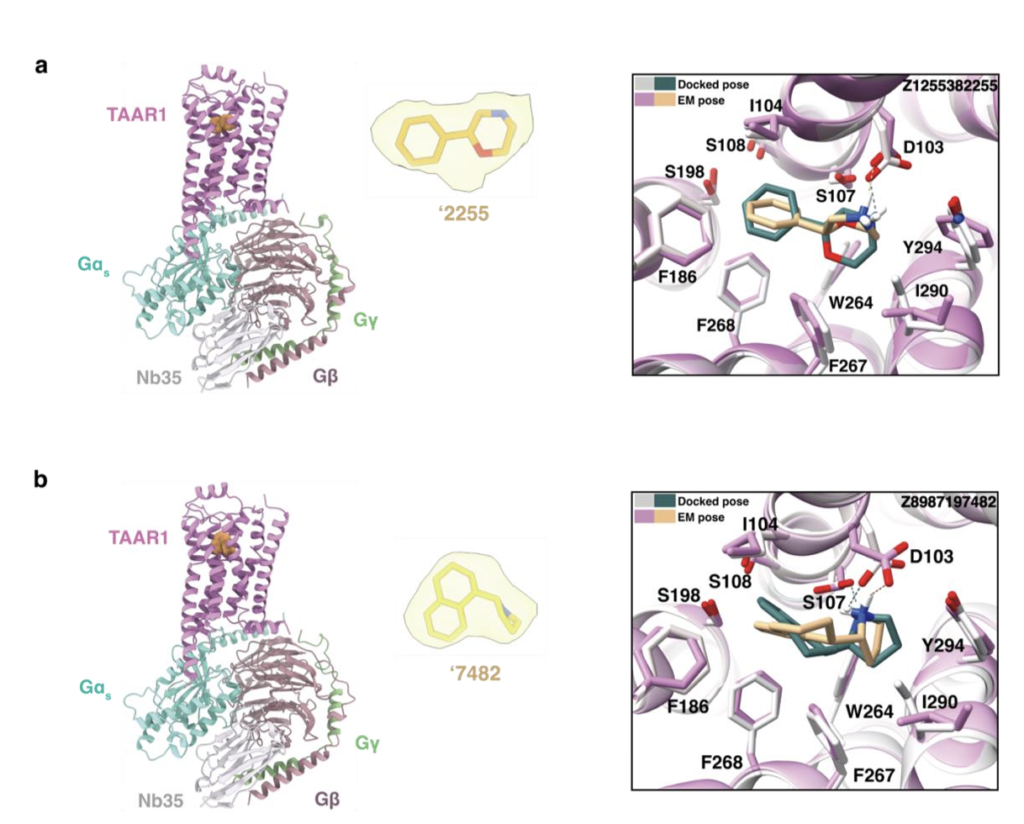
在这两个TAAR1结构中,受体均呈现典型的激活构象,两个激动剂均结合于正构位点,TM6螺旋向外偏移程度相近。TAAR1主要通过两个配体片段识别激动剂:一是芳基片段,与F268⁶・⁵²、F186^(ECL2)、I104³・³³形成的亚口袋发生π-π堆积与疏水相互作用;二是阳离子胺基,与D103³・³² 形成离子对。这些相互作用既被对接预测,也被冷冻电镜结构证实。Z1255382255与Z8987197482的对接预测构象与实验观测构象的RMSD分别为2.3Å与1.4Å(图3a~b)。
尽管实验构象与预测构象形成的受体相互作用一致,但两者结构存在显著差异。Z1255382255的实验构象相较于对接预测的伸展平面构象发生构象旋转,使苯环呈倾斜角度,既与F268⁶・⁵²形成π-π相互作用,又更靠近ECL2环上的F186;同时,饱和环系统内的阳离子氮原子发生位移,优化与D103³・³²的盐桥形成,阳离子氮原子靠近D103³・³²时,该残基略微向外偏移。类似地,Z8987197482向ECL2环偏移,苯环更靠近F268⁶・⁵²,其阳离子氮原子也更靠近D103³・³²,导致该残基的侧向位移大于Z1255382255结合结构(图3a~b)。总体而言,这两个化合物的实验与对接结构共同解释了其构效关系(SAR)。例如,从3nM的吡啶衍生物Z898713207变为1nM的苯环衍生物Z1255382255,与苯环在TAAR1非极性“芳基笼”中的定位一致:吡啶环在该位点需付出去溶剂化代价且无极性相互作用补偿,而苯环则无此代价。同理,Z8700802749(12μM)的吗啉环替换为Z8987197482(48nM)的环己基(两者均结合于非极性亚口袋),后者平面性降低、结合更契合,解释了其效力提升超250倍的原因。
与基于AlphaFold2 TAAR1模型的对接对比
近期一项有趣研究中,研究者采用分子对接技术,针对AlphaFold2(AF2)预测的TAAR1模型发现新型激动剂²⁰。该研究从100个模型中,筛选出能最优富集已知TAAR1配体的模型用于前瞻性大规模库筛选,该策略为该团队首创⁵⁸。实验验证高排名分子后,预测命中率达60%,最优化合物EC₅₀为35nM。从命中率来看,该结果显著优于本研究采用相同对接程序获得的结果,而两者亲和力相近。尽管AF2模型为失活态构象(图4a),但其筛选的所有新配体均为功能性激动剂。如何解释这些现象?
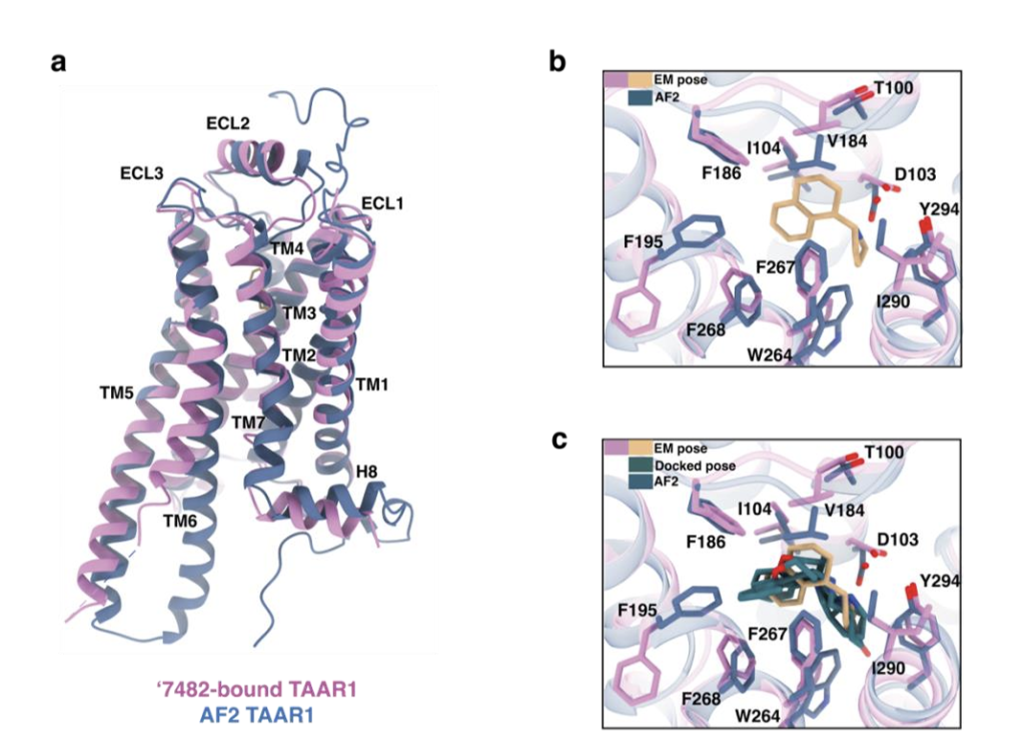
通常认为,实验解析的全激活态结构(如本研究的冷冻电镜结构)应至少与AF2模型效果相当,且更易偏向筛选激动剂。AF2方法的优势之一是计算多个模型,并选择对照实验中配体富集效果最优的模型用于前瞻性筛选,这可能是其前瞻性高命中率的关键,但也可能带来意外后果。AF2模型成功的原因之一,是其正构位点异常紧凑,多个残基向配体口袋收紧,相较于实验结构(图4b),结合口袋体积为513~639ų(因模型而异),远小于本研究实验结构的746ų。这使对接偏向筛选TAAR1偏好的小分子阳离子激动剂,但AF2正构位点过度收缩,导致本研究发现的多种配体无法进入该口袋(图4c)。同时,过度紧凑的口袋也限制了初始分子的效力优化空间,导致AF2对接筛选的最优激动剂优化后效力与初始命中化合物相近。而本研究冷冻电镜结构的更大正构位点,使我们能将化合物优化至1nM水平,最终贡献了其优异的体内效力(见下文)。这表明,通过配体富集选择模型结构的策略(该领域广泛应用,包括本团队研究⁵⁹⁻⁶¹),虽能优化初始发现效率,但可能给后续优化带来挑战。
新型TAAR1激动剂的体内抗精神病样活性
为评估化合物Z8987163207、Z1255382255、Z8987197482的治疗潜力,我们首先测定其在小鼠腹腔注射(10mg/kg)后的药代动力学特征(扩展数据图7、补充数据表2)。三个化合物均具有优异的脑与脑脊液(CSF)暴露量,脑脊液浓度常作为脑内游离浓度(Fu)的通用替代指标。其中Z8987163207的脑脊液峰浓度(Cmax)达3040ng/mL(约18μM),约为Z1255382255与Z8987197482的两倍;脑脊液药时曲线下面积(AUC)为292000ng・min/mL,而Z1255382255与Z8987197482分别为67400与123000ng・min/mL;脑脊液半衰期(T₁/₂)为56分钟,Z1255382255与Z8987197482分别为35与64分钟。综上,Z8987163207、Z1255382255、Z8987197482兼具TAAR1高效力与高脑暴露量,受体覆盖率(游离浓度/EC₅₀)达1000~10000倍。因此,我们通过前脉冲抑制(PPI)行为学实验评估其抗精神病样药效,该实验是公认的TAAR1激动剂药效评价模型¹²,⁶²⁻⁶³。
听觉惊跳反射的前脉冲抑制(PPI)用于评估弱前刺激对后续强惊跳刺激反应的抑制能力。精神分裂症患者存在PPI缺陷⁶⁴,啮齿类动物的类似缺陷被用于模拟精神病样状态⁶⁵。我们通过腹腔注射安非他明(AMPH)诱导小鼠PPI缺陷,该方法是啮齿类PPI破坏模型的经典方案,而抗精神病药物可逆转该缺陷。
小鼠先腹腔注射溶剂(Veh)或新型TAAR1激动剂(图5a~c),10分钟后再注射溶剂或3mg/kg安非他明。实验分组包括:溶剂-溶剂对照组、化合物-溶剂对照组、溶剂-安非他明阳性对照组、化合物-安非他明实验组(统计数据见补充表3,图中显示事后检验值)。Z8987163207实验结果显示,溶剂-安非他明组的PPI相较于溶剂-溶剂对照组与0.5mg/kg Z8987163207-溶剂组显著降低(图5a),而0.5mg/kg Z8987163207可逆转安非他明诱导的PPI 损伤。类似地,Z1255382255实验中,溶剂-安非他明组的PPI相较于溶剂-溶剂对照组、0.2mg/kg Z1255382255-溶剂组、0.2mg/kg Z1255382255-安非他明组显著降低(图5b);0.1mg/kg Z1255382255-安非他明组的PPI相较于溶剂-溶剂对照组与0.2mg/kg Z1255382255-安非他明组降低,表明0.2mg/kg剂量的Z1255382255能更有效逆转安非他明诱导的PPI损伤。最后,Z8987197482实验显示,溶剂-安非他明组的PPI相较于溶剂-溶剂对照组与0.2mg/kg Z8987197482-安非他明组显著受损(p≤0.050),表明0.2mg/kg Z8987197482可有效修复PPI缺陷(图5c)。综上,Z8987163207、Z1255382255、Z8987197482均可逆转溶剂-安非他明诱导的PPI破坏,其中Z1255382255与Z8987197482效果更优,可能与其靶点效力更高相关。三个化合物均能强效改善安非他明小鼠模型的PPI缺陷。
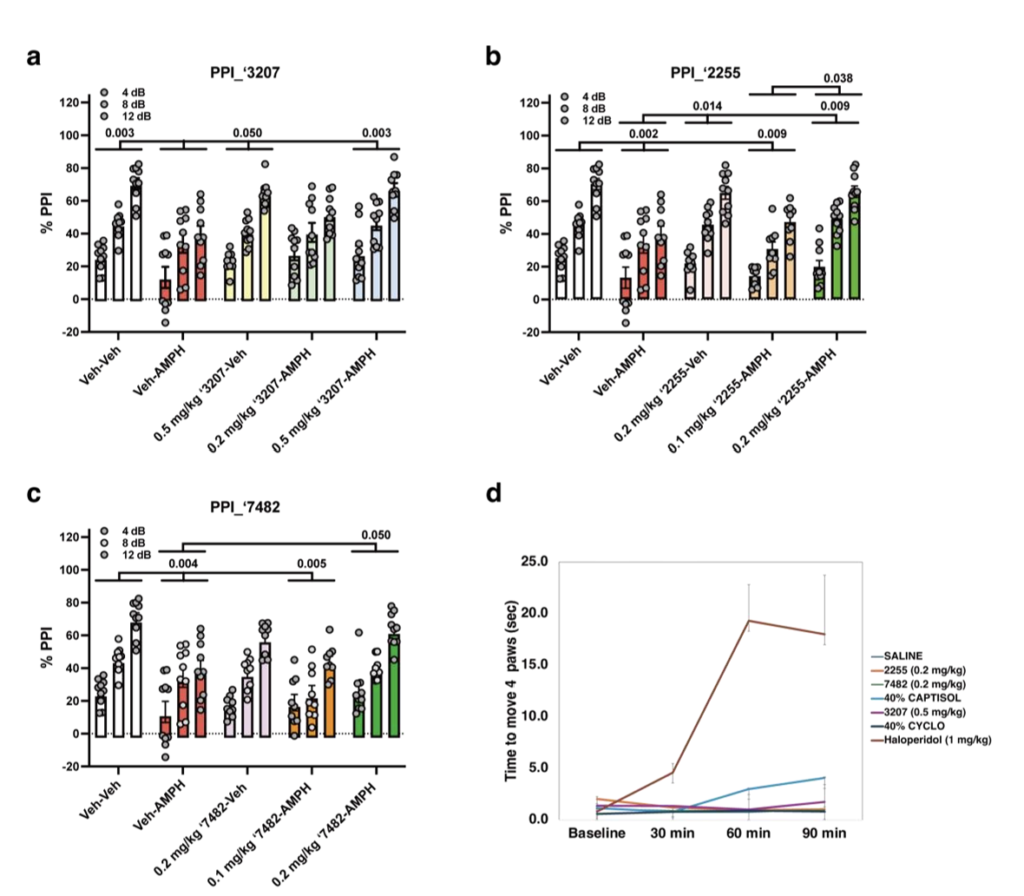
除PPI外,我们还评估了三个激动剂对空白反应、惊跳反应与僵直行为的影响。溶剂-溶剂组与化合物-溶剂组的空白反应持续处于低水平,显著低于溶剂-安非他明组与至少一个化合物-安非他明组(补充图2a~c)。三个化合物对脉冲/惊跳反应的影响略有差异,但总体而言,各化合物最高剂量组的脉冲反应低于其他组(补充图2d~f)。我们还评估了化合物诱发僵直的潜力,这是当前多数抗精神病药的严重不良反应。在能逆转安非他明诱导PPI破坏的剂量下,Z1255382255(0.2mg/kg,腹腔注射)、Z8987197482(0.2mg/kg,腹腔注射)、Z8987163207(0.5mg/kg,腹腔注射)处理的小鼠,在60分钟与90分钟均未观察到僵直行为;而经典多巴胺受体拮抗剂氟哌啶醇(1mg/kg,腹腔注射)处理的小鼠则出现明显僵直,且持续至少90分钟(图5d)。这表明,新型激动剂相较于经典抗多巴胺能药物,具有更优的僵直相关安全性。基于小鼠PPI实验的文献数据,新型化合物在低至0.2mg/kg剂量下即展现显著活性,其效力至少与乌洛塔罗特相当,甚至更优。
4
结论
本研究有四项核心发现值得重点强调:第一,通过大规模分子对接与结构导向优化,获得了对TAAR1具有纳摩尔级效力、对5-HT₁A具有亚微摩尔级活性的激动剂,其体外与小鼠体内效力均优于乌洛塔罗特。第二,筛选仅获得激动剂,这可能源于对接采用TAAR1激活态结构——其正构位点收缩包裹激动剂,优先选择小分子激动剂样化合物,排斥大分子拮抗剂样化合物。该观点与此前基于AlphaFold2 TAAR1模型的对接研究一致²⁰:尽管AF2模型整体为失活态,但其正构位点收缩更显著,激动剂命中率更高。AF2模型过度紧凑的正构位点虽提升命中率,但可能限制后续强效先导化合物优化,这可能是优化对接参数时过度适配AF2模型结构所致。第三,本研究的对接预测结果基本被后续冷冻电镜结构验证,为化合物进一步优化提供模板。第四,三个新型激动剂能强效逆转安非他明诱导的 PPI 破坏,且不引发经典多巴胺受体拮抗剂的关键副作用——僵直。这些新型TAAR1激动剂相较于已上市的典型抗精神病药,可能具有更宽的治疗指数。
本研究存在若干局限性:尽管获得了对TAAR1与5-HT₁A受体效力提升的激动剂,但其主要靶点仍为TAAR1,对5-HT₁A仅具中等效力。新型激动剂虽与已知TAAR1配体拓扑结构不同,但因受体正构位点狭小,共享部分物理特征与部分核心骨架。最后,仅通过安非他明诱导的急性精神病PPI模型评估了Z8987163207、Z1255382255、Z8987197482的体内活性,其对其他行为表型的影响及潜在不良反应尚未探究。
这些局限性并未掩盖本研究的核心成果:针对TAAR1的大规模分子对接结合结构优化,发现了一系列与已知配体拓扑结构无关的低纳摩尔级TAAR1激动剂。靶向受体激活态成功偏向筛选激动剂,冷冻电镜结构基本验证了对接预测。新型TAAR1激动剂的优异物理性质(库选择与对接命中时严格把控)使其具备高中枢神经系统暴露量,行为学改变既改善感觉运动门控,又降低僵直副作用。这些化合物的结构与药理学特性区别于已知TAAR1在研药物,支持其进一步优化,同时也验证了基于结构的配体发现策略在该重要受体研究中的价值¹⁷,²⁰。
文中全新化合物及其二维结构列表见补充表1~2,所有化合物可从Enamine订购。