1、摘要
本文公开了一种稳健且可规模化的合成平台,可广泛获取噻丁烷类砌块,覆盖硫的三种氧化态(S(II)、S(IV)和S(VI))。以易获取的噻丁烷-3-酮或2-(氯甲基)硫杂丙环为起始原料,制备了一系列多样化的3-取代和3,3-二取代噻丁烷,包括羧酸、伯胺、仲胺、醇、肼、烷基卤化物、磺酰卤化物等。通过合成上市N-酰基磺酰胺药物的噻丁烷类似物,证实了这些片段的合成实用性。此外,通过与氧杂环丁烷、氮杂环丁烷和环丁烷类似物的直接对比,对新型噻丁烷砌块的理化性质进行了系统研究。胺类和羧酸类的pKa数据,以及模型苯甲酰胺和苯胺类的LogD测定结果,揭示了依赖于硫原子氧化态的明确构效关系。S(II)噻丁烷衍生物的亲脂性与环丁烷接近,而S(IV)和S(VI) 衍生物的亲脂性显著更低,酸性更强/碱性更弱,极性甚至超过氧杂环丁烷。这些发现支持噻丁烷作为多功能“三合一”片段的理念——通过调节硫的氧化态可精细调控电离态和亲脂性,为现代药物发现提供富含sp³杂化碳、极性可调的砌块合理设计方案。
2、引言
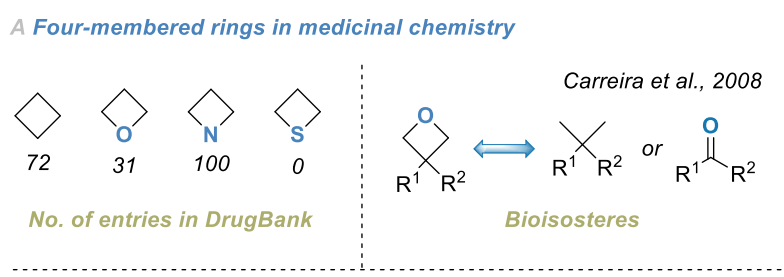
过去二十年来,四元环在药物发现中的应用显著增加[1]。事实上,环丁烷、氧杂环丁烷和氮杂环丁烷已受到合成化学家和药物化学家的广泛关注(图1A)[2-10]。其中,氧杂环丁烷的发展尤为引人注目:经Carreira团队的开创性研究提出其可作为羰基和偕二甲基片段的生物电子等排体替代物后(图1B)[10,11],众多学术和工业团队纷纷投身该领域研究[4-8,12-14],最终催生了多个药物候选物。
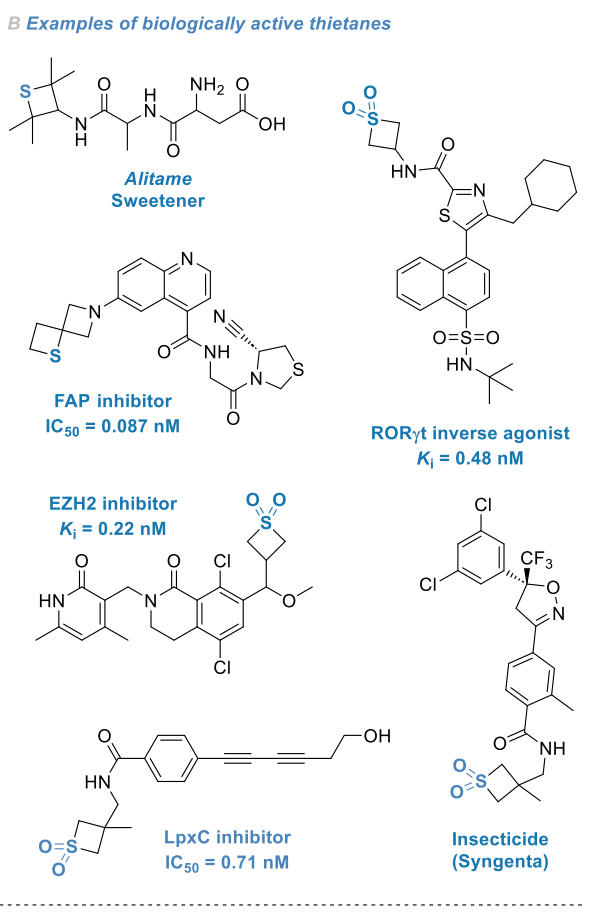
相比之下,噻丁烷在药物发现中却未得到足够重视[6,15]。尽管如此,文献中已报道多种含该杂环的生物活性分子,包括二肽甜味剂阿力甜[16]、视黄酸受体孤儿受体γt亚型(RORγt)反向激动剂[17]、zeste同源物2增强子(EZH2)抑制剂[18]、脯氨酰内肽酶成纤维细胞活化蛋白(FAP)抑制剂[19]、核苷类似物[20-22]及农药[23](图1B)。
一般而言,噻丁烷的反应活性略低于氧杂环丁烷或氮杂环丁烷,这归因于硫原子电负性较低、原子半径较大,导致环张力更小[24]。因此,噻丁烷稳定性更高——与含氧化合物和含氮类似物相比,其在酸性介质或与强亲核试剂反应时更不易发生开环反应[13,25]。
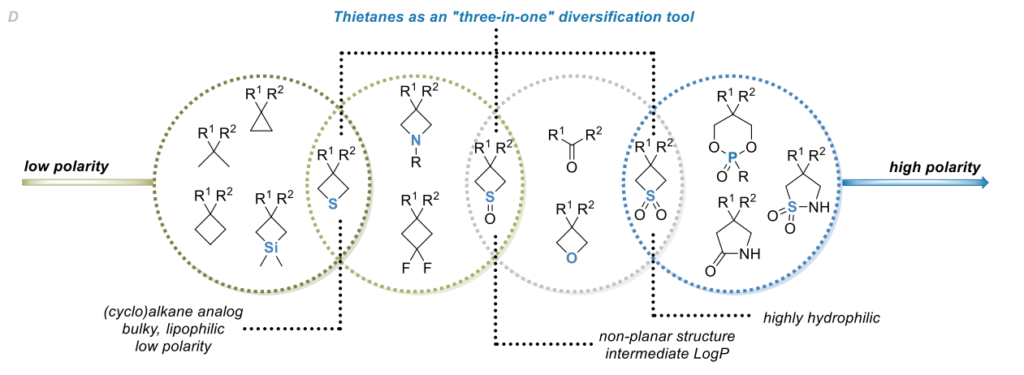
与其他饱和四元杂环不同,噻丁烷可发生氧化还原反应。特别是杂环开环可通过催化脱硫实现[26,27],而硫原子的氧化转化更为重要——可获得理化性质显著改变的系列衍生物(即四元环亚砜和砜)。此类修饰能显著调节化合物的亲脂性和酸碱性质,使噻丁烷成为一种可调谐的“三合一”多样化工具,可模拟药物化学中多种常用结构片段[28](图1D)。因此,已有多个团队提出将不同硫氧化态的噻丁烷用作氧杂环丁烷类似物[15,23,29]。
噻丁烷衍生物的合成方法早已见诸报道:早在1966年就有首篇综合综述发表[30],此后又有多篇更新[31,32]。常规合成方法包括硫醚二阴离子与1,3-双亲电试剂的双烷基化[33]、烯烃与硫羰基化合物的光化学thiaPaternò-Büchi [2+2]环加成[34-36],或三元杂环的重排反应[31]。近期研究报道了对已含噻丁烷片段化合物的修饰——例如Bull团队报道的噻丁烷1,1-二氧化物合成[23]。
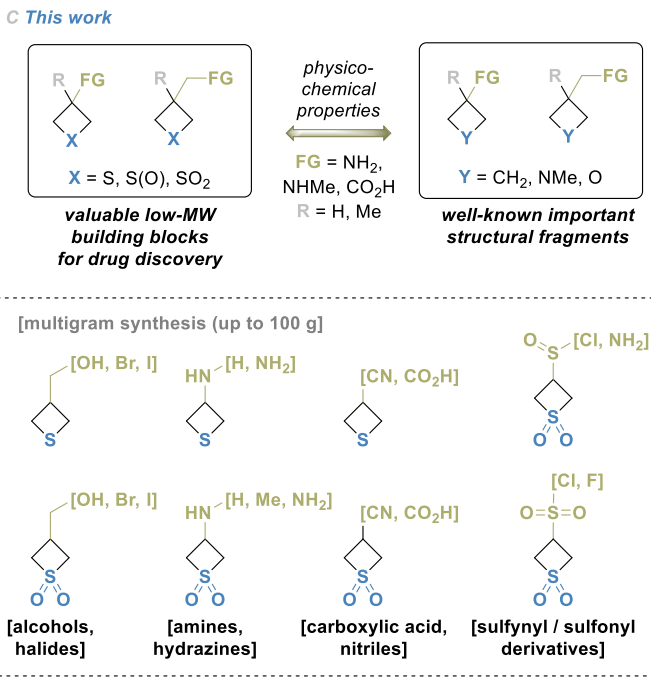
本研究中,我们报道了一系列低分子量3-取代噻丁烷砌块的制备(图1C)[37,38]。需要强调的是,简单功能化噻丁烷的合成极具挑战性,因其物理性质特殊(挥发性和/或高亲水性),相关文献报道极少。本研究之前,几乎没有可规模化合成的可靠方案,多数目标化合物甚至缺乏光谱表征数据。为推动功能化噻丁烷在药物发现等领域的广泛应用,本文详细描述了图1C所示系列砌块的合成及完整表征。我们特意采用简单、稳健且成熟的转化反应,既保证方法的高效性,也验证噻丁烷环对有机化学中常见反应的耐受性。此外,我们系统表征了不同硫氧化态模型噻丁烷衍生物的理化性质(即pKa和LogD),并与其他四元环类似物对比,通过实验证实图1D所示理念。最后,通过虚拟库类先导化合物性质评估及经典抗菌药物磺胺醋酰类似物的合成,进一步验证了所合成砌块在药物化学中的实用性。
3、结果与讨论
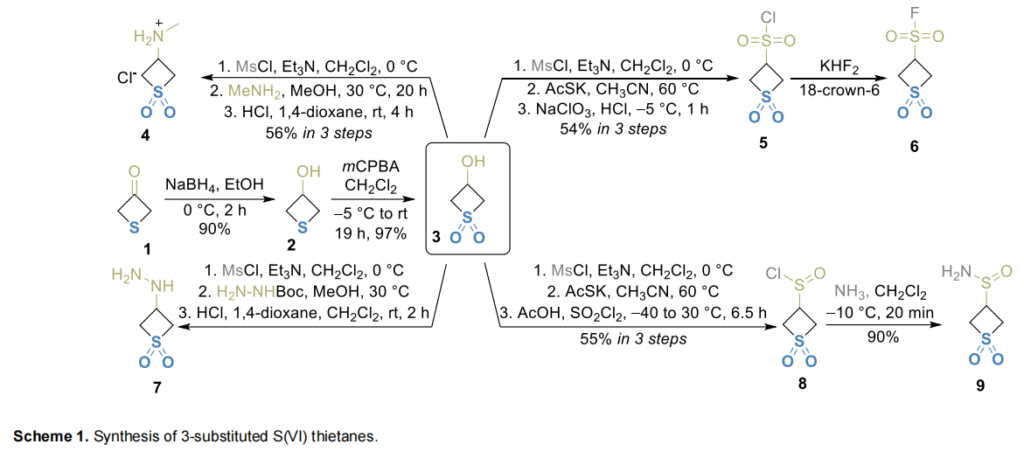
噻丁烷-3-酮(1)是目前最易获取的噻丁烷类试剂之一,为制备3-取代噻丁烷衍生物提供了丰富的反应位点。该起始原料可千克级采购,是合成含S(II)、S(IV)和S(VI)氧化态硫原子噻丁烷砌块的便捷原料。酮1经硼氢化钠还原得到醇2,随后用间氯过氧苯甲酸(mCPBA)氧化生成砜3(Scheme 1)。3经甲磺酰化后发生亲核取代反应,得到N-甲基胺4和肼7;甲磺酰化产物还可转化为硫代乙酸酯,进而作为前体经酸性条件下的氯酸钠氧化制备磺酰氯5和磺酰氟6;用磺酰氯进行温和氧化可得到亚磺酰氯8,进而制备亚磺酰胺9。
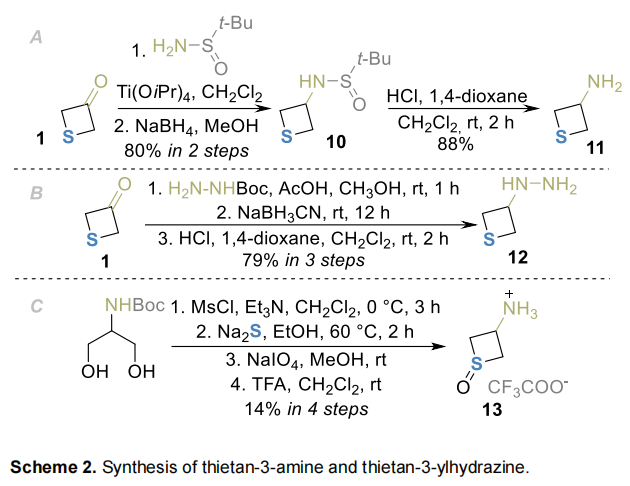
采用替代路线从酮1制备含S(II)的噻丁烷含氮衍生物:通过Ellman's亚磺酰胺还原胺化高效制备胺11(Scheme 2A),用叔丁氧羰基肼制备肼12(Scheme 2B)。此前胺11通过无环二醇环化制备,但该方法难以规模化且产物纯度低;对该方法进行轻微修饰(加入高碘酸钠介导的氧化),经选择性氧化和N-叔丁氧羰基(N-Boc)脱保护,成功合成相应亚砜13(Scheme 2C)。
第三条合成路线以2-(氯甲基)硫杂丙环(14)为原料:与氰化钾反应生成腈15,经硫氧化得到砜16(Scheme 3);腈15经氢化铝锂(LiAlH₄)还原得到胺17,经N-叔丁氧羰基保护、氧化和脱保护序列,得到相应氧化类似物18;腈经碱性水解得到羧酸19,可进一步转化为相应酯20;用间氯过氧苯甲酸氧化噻丁烷前体19,通过调节试剂用量控制氧化程度——1.1当量间氯过氧苯甲酸得到亚砜21(S(IV)),过量氧化剂得到砜22(S(VI))。
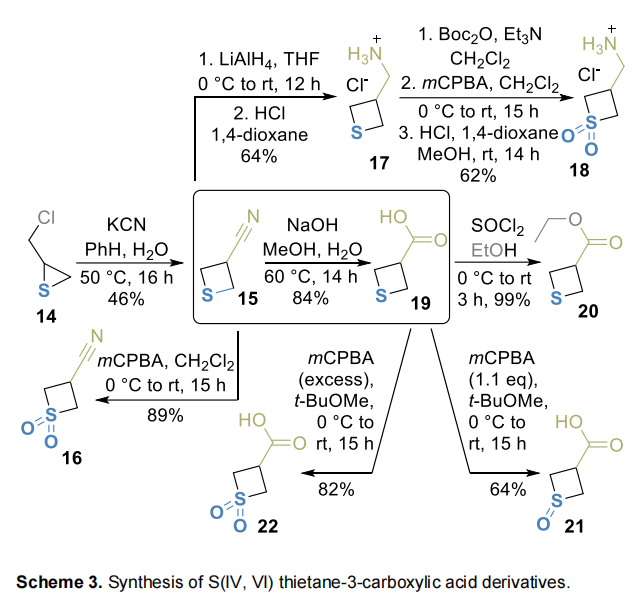
羧酸19的制备为合成相应同源醇23提供了可能:羧酸19经硼烷(BH₃)还原得到醇23,再用间氯过氧苯甲酸氧化生成砜24;醇经甲磺酰化和卤离子亲核取代反应,制备溴化物25和碘化物26(潜在烷基化试剂);参照化合物4的制备方法,从醇24可轻松得到N-甲基胺27(Scheme 4)。
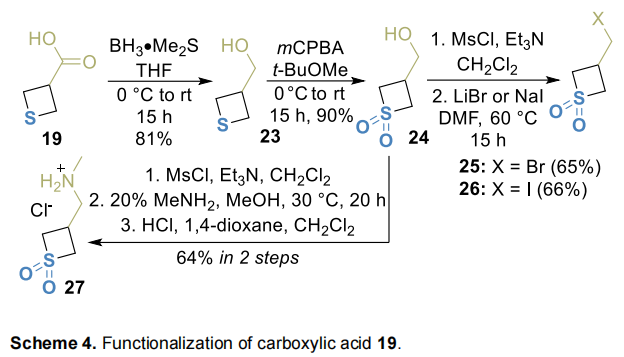
下一步合成同源物(如醇29):将羧酸19转化为Weinreb酰胺,与Grignard试剂加成得到酮28,以此为前体制备仲醇的S(II)和S(VI)衍生物29、30及同源胺31、32(Scheme 5)。
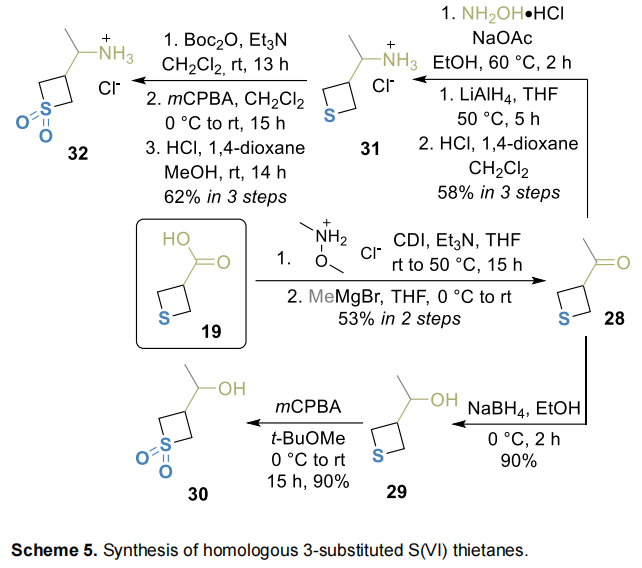
以起始酮1为原料,通过稳定叶立德的维蒂希反应得到相应α,β-不饱和酯33(经硫氧化得到衍生物34)和丙烯腈35;丙烯腈35经碳-碳双键还原得到36,水解后得到噻丁基乙酸37,再氧化生成砜38;腈36经氢化铝锂还原得到胺39,采用上述N-保护、氧化和脱保护方法得到其S(VI)氧化类似物40(Scheme 6)。
同源物37经标准衍生化制备系列砌块:经硼烷还原得到醇41,氧化生成相应砜42,进而用于制备溴化物43 和磺酰氯44(Scheme 7)。
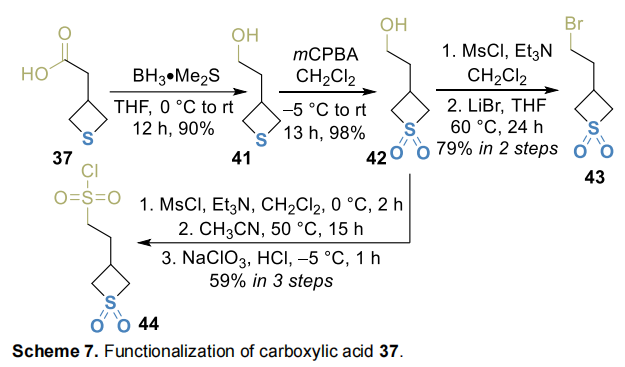
我们进一步探索了高效制备3,3-二取代噻丁烷的可能性:腈15经去质子化-烷基化序列在C(3)-位引入甲基,得到45;经水解和库尔修斯重排分别得到羧酸46和相应伯胺47(Scheme 8)。
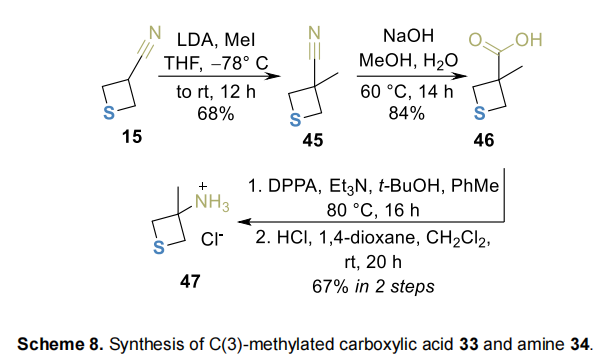
提出一种步骤更简洁的替代合成路线:氧杂环丁烷经溴化氢开环中间体,再经分子内硫烷基化得到同源醇50;经甲磺酰化-亲核取代反应序列制备叠氮51,再经硫氧化得到相应砜52;催化氢化顺利得到胺53(Scheme 9)。
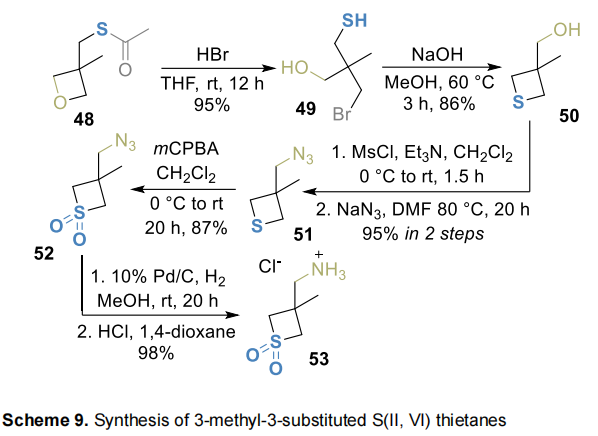
醇50经两步氧化得到同源S(VI)羧酸55,经改进的叠氮磷酸二苯酯(DPPA)介导的库尔修斯重排,酸性后处理得到相应胺盐酸盐56(Scheme 10);尝试参照胺11的制备方法通过二醇环化合成胺41,但无论何种条件,目标产物收率均不理想。
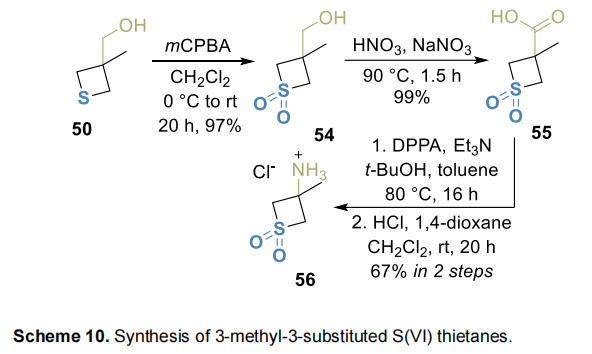
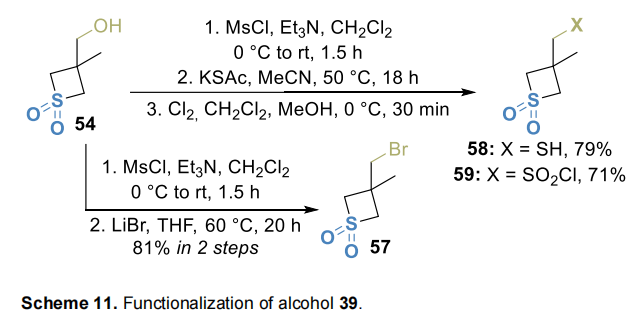
醇54成功用于制备溴化物57、硫醇58和磺酰氯59(Scheme 11)。
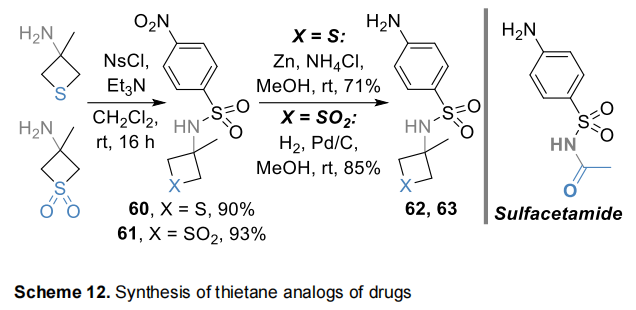
为验证所提砌块的实用性,我们制备了上市药物的类似物。如前所述,酰基磺酰胺基团中的羰基可被四元杂环有效替代[39]。因此,我们选择经典抗菌药物磺胺醋酰为模型,设计并合成其电子等排类似物62和63:胺47和56均与对硝基苯磺酰氯(nosyl chloride)发生磺化反应,根据硫原子氧化态不同,分别用锌/氯化铵(S(II)噻丁烷)或钯催化氢化(噻丁烷二氧化物)还原硝基(Scheme 12)。
以所得羧酸和胺为基础,研究噻丁烷衍生物的理化性质,并选择相应氧杂环丁烷、N-甲基氮杂环丁烷和环丁烷作为模型化合物进行对比。测定所有上述胺类和羧酸类的pKa值(图2),以及模型衍生物(母体羧酸的苯胺类和胺与苯甲酰氯(BzCl)反应得到的苯甲酰胺类)的LogD值(图3,实验细节见支持信息)。
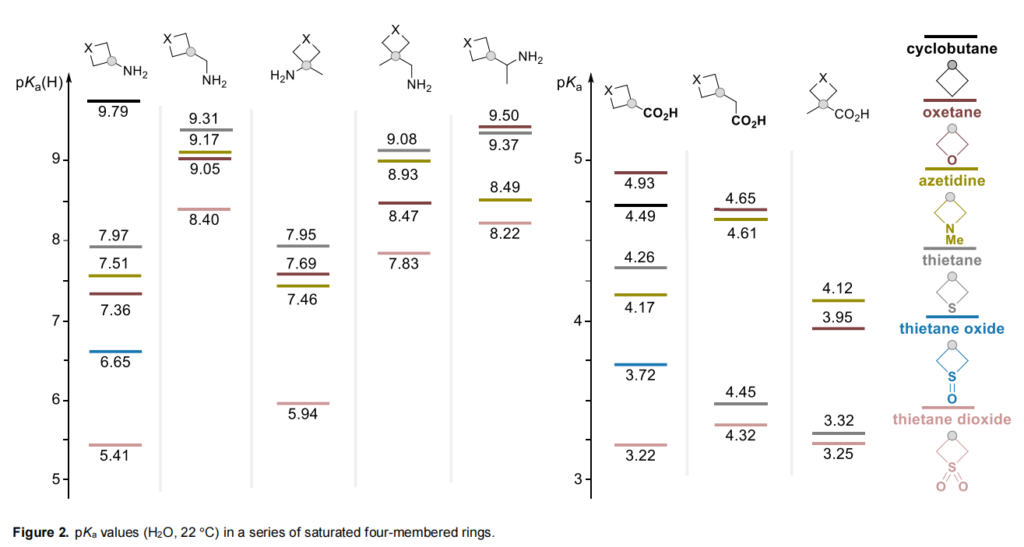
pKa数据分析表明,对于伯胺类,所有杂环类似物的碱性均显著低于环丁烷衍生物(pKa=9.79)。对比同源胺R-NH₂和R-CH₂NH₂的数据发现,除杂原子的诱导效应外,杂环的构象行为可能是重要影响因素;对于R-CH₂NH₂系列,杂原子的影响仍显著,但所有化合物的碱性差异大幅减小。所有代表物中碱性最弱的是噻丁烷二氧化物(pKa=5.41),从R-CH₂NH₂转变为R-NH₂时,其碱性降幅超过2个单位;噻丁烷monoxide的pKa值(6.65)也低于伯胺的预期值。其他化合物的碱性:R-NH₂系列为7.0-8.0,R-CH₂NH₂系列为8.5-9.5;除氨乙基取代物外,碱性最强的衍生物含噻丁烷环,这归因于硫原子的诱导效应可忽略。N-叔丁氧羰基氮杂环丁烷和氧杂环丁烷衍生物的pKa值处于相近范围,无特定相关性。
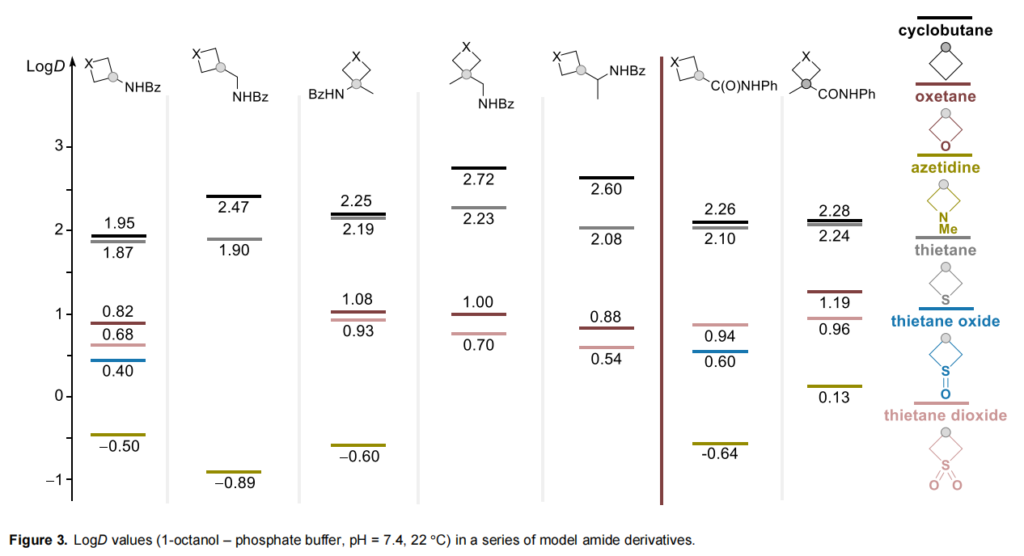
羧酸系列结果类似:正如预期,氧化噻丁烷衍生物酸性最强(砜类pKa=3.22,亚砜类pKa=3.72);令人意外的是,氧杂环丁烷羧酸的平均酸性(pKa=3.9-4.9)低于噻丁烷衍生物;其余羧酸的pKa=3.5-4.5,均低于环丁烷羧酸的相应值。从噻丁烷羧酸转变为相应亚砜和砜时,pKa值下降0.5个单位,而同源羧酸的酸性差异极小。采用所有所得杂环的模型苯甲酰胺和苯胺类研究亲脂性:这些衍生物电离效应减弱,可高效用于串联质谱定量分析,具有稳定且易检测的碎片离子,且LogD值处于0-3.5的适宜范围(小分子分析所需);由于碱性原因,N-甲基氮杂环丁烷的LogD值大多为负值。
本研究的核心预期得到充分证实:噻丁烷单氧化物是所有环中亲脂性最低的(LogD=0.4),显著低于以高亲水性著称的氧杂环丁烷;其原因是亚砜基团的偶极矩显著,与官能团偶极相互作用,且溶剂化效应更有利。噻丁烷二氧化物的亲脂性(LogD=0.7)也低于氧杂环丁烷(LogD=0.8),但高于相应亚砜;S(II)噻丁烷的亲脂性(LogD=1.87)与环丁烷(LogD=1.95)略有差异。这些结果证实:含S(II)的噻丁烷可作为(环)烷烃类似物,而氧化噻丁烷可作为饱和杂环系统的极性模拟物。所有研究衍生物均呈现以下亲脂性顺序:环丁烷<噻丁烷<<氧杂环丁烷<噻丁烷S,S-二氧化物<噻丁烷S-monoxide<N-甲基氮杂环丁烷。
所得结果表明,使用不同硫氧化度的噻丁烷可有效调节物质的理化性质。例如,可获得比氧杂环丁烷亲脂性更高的S(II)类似物,以及亲脂性更低的S(IV)、S(VI)类似物,且可调节分子中相应位点的酸性或碱性。
4、结论
本文公开了一种可规模化的合成平台,用于制备涵盖硫三种氧化态(S(II)、S(IV)和S(VI))的系列噻丁烷类砌块。以易获取的噻丁烷-3-酮和2-(氯甲基)硫杂丙环为起始原料,通过一系列稳健高效的官能团转化反应,获得了多样化的3-取代和3,3-二取代噻丁烷。
所得库包含同源和异构羧酸、伯胺、仲胺、肼、醇、烷基卤化物、亚磺酰氯、磺酰氯、磺酰氟及相关衍生物,所有化合物均适用于进一步多样化修饰(本研究已充分证实)。
此外,磺胺醋酰噻丁烷代表类似物的成功合成,证实了这些砌块在药物相关骨架后期修饰中的实用价值。
通过与氧杂环丁烷、N-甲基氮杂环丁烷和环丁烷类似物的直接对比,对所得噻丁烷及其氧化同系物进行系统pKa和LogD研究,揭示了明确的构效关系。噻丁烷环中硫原子的氧化导致胺类碱性可预测性降低、羧酸类酸性升高,其中亚砜和砜是系列中酸性最强、碱性最弱的代表物;同时,噻丁烷S-monoxide和S,S-二氧化物衍生物的亲脂性低于氧杂环丁烷,而S(II)噻丁烷的亲脂性与环丁烷接近。
这些趋势证实:S(II)-噻丁烷表现为烷烃样片段(如环丁烷——许多生物活性化合物中重要且常用的结构片段),而其氧化同系物作为极性更高的类似物,可在单一化学型内精细调节电离态和亲脂性。
因此,噻丁烷尽管在药物化学中长期被忽视,但因其可通过改变硫原子氧化态精细调节理化性质,成为极具吸引力的结构基序。这种多功能“三合一”四元环片段可模拟多种常见结构基序(从(环)烷烃到其他杂环)。我们预期这些发现及所描述的片段将在药物发现项目中得到广泛应用。
本文中涉及的含噻丁烷砌块与相关衍生物,Enamine 均有现货供应。