摘要
含氟饱和杂环是药物设计中极具价值但尚未被充分探索的结构单元。本文提出了一种可规模化的合成策略,以简单易得的起始原料合成含氟四氢呋喃(THF)和四氢噻吩(THT)构建块。优化后的反应条件能够以十克级规模制备三氟甲基(CF₃)取代酮类化合物,以及相应的立体结构明确的醇、胺和氨基酸衍生物。理化性质分析揭示了清晰的构效关系:杂原子和亚砜(SO₂)基团的强诱导效应会影响化合物的酸解离常数(pKa)和等电点(pI),其中三氟甲基取代可显著增强化合物的酸性并降低等电点;亲脂性(LogP)测定结果表明,亲脂性依赖于骨架结构和取代基类型,含硫结构单元的亲脂性最强,而砜类衍生物的亲脂性最弱。总之,本研究为获取多样化的含氟砌块(包括非天然氨基酸)提供了一个可靠的平台,有望应用于药物研发领域。
1、引言
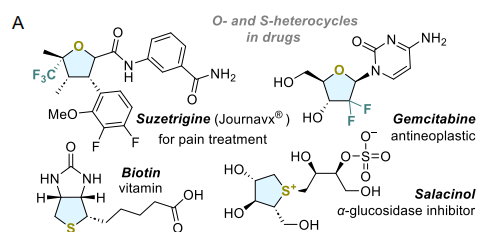
含氧化合物或硫原子的五元杂环(如四氢呋喃和四氢噻吩)是化学和生物学领域中具有优势的结构单元。这些饱和杂环广泛存在于天然产物中,尤其是海洋来源的天然产物,它们具有多种生物功能,包括酶抑制、抗菌活性等,同时也存在于美国食品药品监督管理局(FDA)批准的合成药物中(图1A)[1,2]。代表性例子包括生物素(维生素B₇)[3]、强效α-葡萄糖苷酶抑制剂Salacinol[4]、天然抗菌药物白霉素[5]、二聚倍半萜硫生物碱家族[6]、聚醚类海洋抗生素[7]以及乙酰精宁[8]。除天然产物外,吡啶取代的四氢噻吩已被报道具有抗溃疡和抗分泌活性[9]。临床上使用的含四氢呋喃结构药物包括吉西他滨(及其类似物巯基胞苷)、索磷布韦和西达祖定[10],而最近批准的Nav1.8抑制剂Journavx作为首个用于急性疼痛管理的非阿片类镇痛药,充分体现了该结构单元的药理学重要性[11,12]。
尽管这类结构应用广泛,但四氢呋喃和四氢噻吩单元在药物研发中的进一步应用常受到其代谢稳定性有限的限制。杂原子α位的C-H键是公认的代谢弱点,容易发生快速氧化降解[13]。通过引入氟原子或氟烷基取代基来阻断这些位点,已被证实是调节目标生物活性的有效策略[14-17]。在含氟基团中,三氟甲基(CF₃)取代基尤为重要,它能同时提高代谢稳定性、亲脂性和生物利用度,是药物化学中最强大的设计工具之一[18-21]。
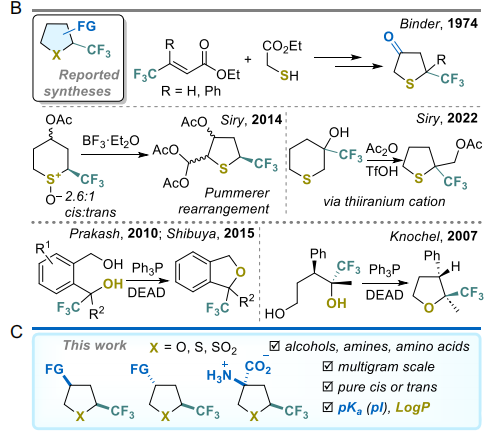
然而,α-三氟甲基取代四氢呋喃和四氢噻吩衍生物的合成方法仍然有限且具有特异性(图1B)。对于四氢噻吩衍生物,已报道的主要合成策略有两种:(a)以无环含硫和含氟前体构建环结构,该方法由Binder及其同事首创[22,23],随后其他研究小组将其发展为有机催化版本[24-26];(b)六元环前体的环收缩和重排反应[27-29]。与此同时,α-三氟甲基取代四氢呋喃衍生物的合成主要通过不同来源的二醇分子内Mitsunobu环化反应实现[30-32]。
本研究报道了一种高效的多克级合成方法,用于制备非对映纯的3-官能化5-三氟甲基取代四氢呋喃和四氢噻吩衍生物(包括胺、醇和氨基酸)——这些化合物是药物化学中极具潜力的砌块。此外,我们还对所选代表性化合物或其模型衍生物进行了理化性质分析,包括酸碱性质(pKa和等电点pI)和亲脂性(LogP)表征,以阐明标题含氟骨架作为未来药物研发工具的潜力。
2、结果与讨论
2.1 合成
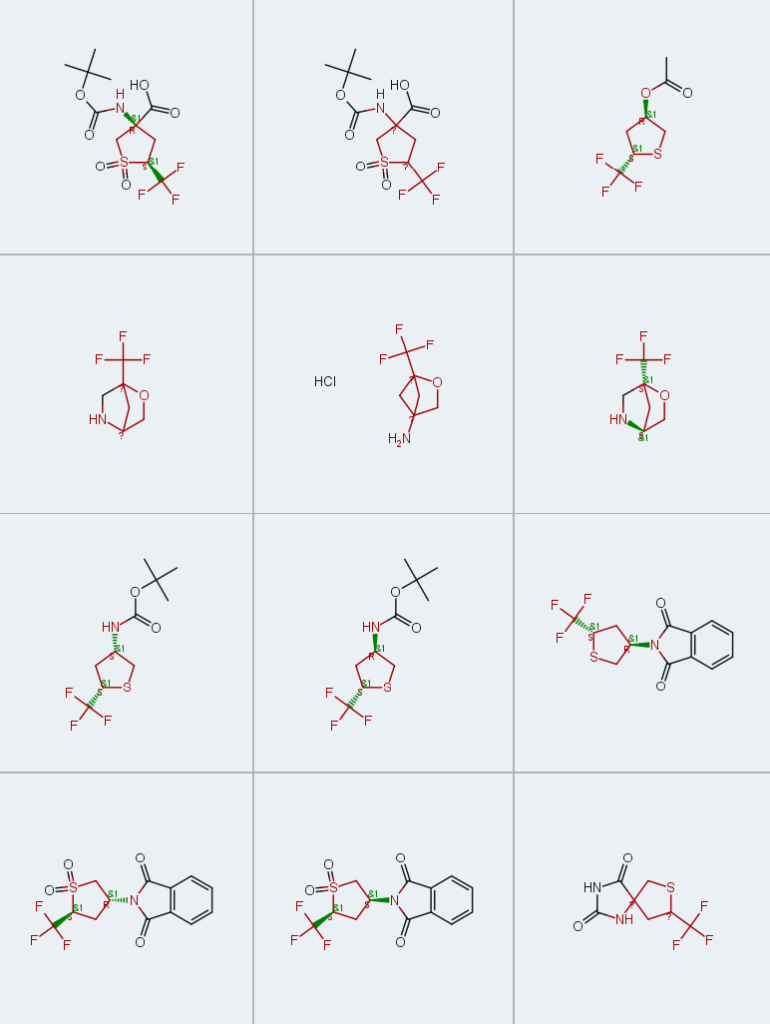
在已报道的合成策略中,Binder及其同事开发的合成5-三氟甲基取代二氢噻吩-3(2H)-酮的方法[22,23],因其简便性和合成吸引力而备受关注。我们对原始工作中提及的最简单酮类化合物1a的制备方案进行了优化(方案1)。用亲核性更低、挥发性更强的吡啶替代哌啶作为硫醇2a与丙烯酸酯3加成反应的介质,显著简化了产物分离过程,无需萃取后处理即可获得近乎定量产率的中间体4。进一步的方法优化(即使用甲醇钠替代乙醇钠,以及用1M硫酸替代浓硫酸对中间体5a进行脱羧反应)共同将产物1a的总产率从三步66%提高至77%,并实现了反应的高可扩展性(由200g原料3可制备多达158g产物1a)。
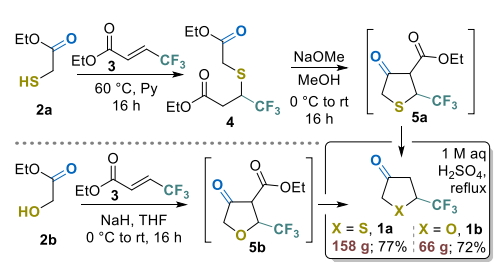
尽管类似方法已用于制备中间体5b的非氟代(甲基取代)类似物[26],但将该方法应用于由乙醇酸乙酯(2b)和酯3合成5-三氟甲基酮1b时未取得成功。在我们的实验中,使用氢化钠(NaH)作为碱有利于高效的迈克尔加成反应。此外,使用稍过量的碱可在同一步骤中实现分子内环化,无需分离迈克尔加成中间体,即可通过一锅法获得酮酯5b。经水解和脱羧反应后,以72%的总产率和高达66g的规模获得了此前未报道的酮类化合物1b。
通过对酮1a和1b中羰基的顺式立体选择性还原,实现了具有明确相对构型的构建块的合成(方案2)。幸运的是,使用易得的硼氢化钠(NaBH₄)即可成功实现选择性还原,以高分离产率获得纯立体异构体形式的相应仲醇顺式-6a和顺式-6b。其他非对映异构体反式-6a和反式-6b通过Mitsunobu构型翻转反应制备[33]。顺式-6a和反式-6a的相对构型通过核Overhauser效应(NOE)实验得到证实(方案2)。
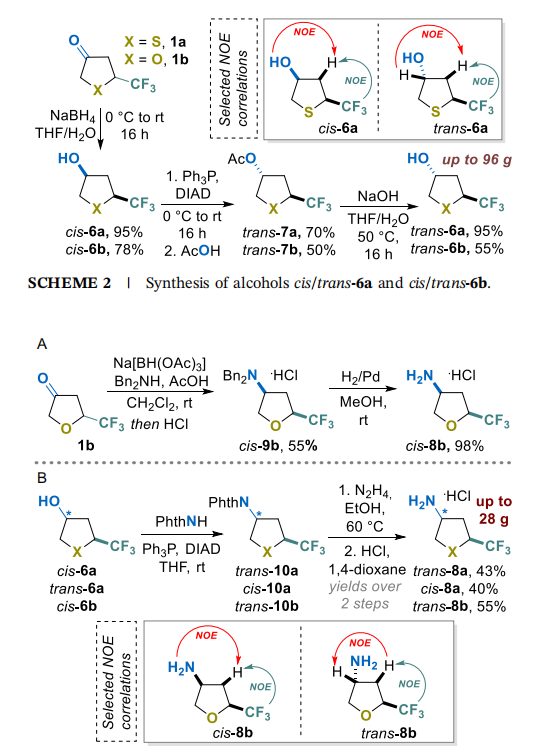
相应胺盐酸盐顺式/反式-8a和顺式/反式-8b的合成过程相对复杂。使用三乙酰氧基硼氢化钠 [34]对酮1b与二苄胺进行还原胺化反应,以55%的满意产率得到胺顺式-9b(方案3A)。随后,通过催化脱除其盐酸盐中的苄基,以十克级规模和98%的产率获得了伯胺盐酸盐顺式-8b。显然,由于硫醚片段的存在,该方法不适用于化合物顺式-8a的合成。
另一种可制备其余三种胺(顺式/反式-8a和反式-8b)的方法,基于醇顺式/反式-6a和顺式-6b与邻苯二甲酰亚胺作为亲核试剂的Mitsunobu反应(方案3B)。正如预期的那样,获得了相应的N-保护中间体反式/顺式-10a和反式-10b,且在C-3位发生了构型翻转[35]。随后,通过脱除邻苯二甲酰亚胺基团,以良好的总产率(三步40%-55%)和高达28g的规模获得了伯胺反式-8a、b和顺式-8a。顺式-8b和反式-8b的相对构型通过NOE实验得到证实。
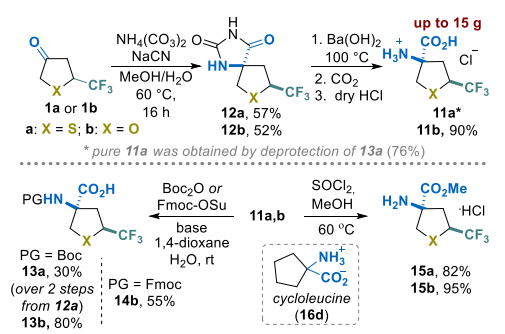
季铵氨基酸11a和11b(盐酸盐)的合成基于酮1a和1b的Bucherer−Bergs反应[36,37](方案4)。以纯非对映异构体形式获得了相应的乙内酰脲12a和12b,产率分别为57%和52%。通过碱性水解反应完成合成[38-40]。然而,含氧杂环氨基酸11b的分离无需额外纯化,而我们尝试直接从乙内酰脲中获得纯11a样品的所有努力均未成功。为解决这一问题,将粗品化合物11a转化为相应的N-Boc保护衍生物13a(两步分离产率30%),然后通过酸性裂解保护基,从乙腈(CH₃CN)中重结晶后获得目标氨基酸盐酸盐11a(产率76%)。11b的氨基可通过Boc或Fmoc保护基进行保护,分别以55%和80%的产率获得砌块14b和15b。此外,盐酸盐11a和11b可高分离产率转化为相应的酯16a和16b。
所制备的氨基酸衍生物可视为环亮氨酸(16d)的类似物,环亮氨酸是一种非蛋白原性、代谢稳定的氨基酸,广泛应用于生物医学研究[41,42]。
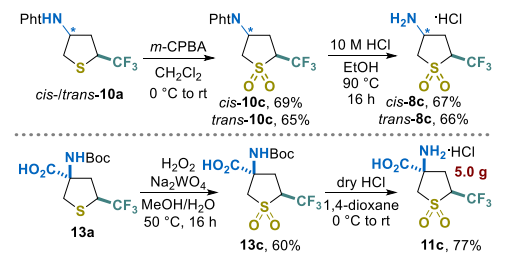
最后,我们验证了所合成构建块用于制备官能化四氢噻吩砜的可行性。因此,使用间氯过氧苯甲酸(m-CPBA)[43,44]对N-邻苯二甲酰亚胺衍生物顺式-10a和反式-10a中的硫原子进行氧化反应,反应顺利进行,分别以69%和65%的产率获得相应中间体顺式-10c和反式-10c(方案5)。随后,通过邻苯二甲酰亚胺部分的酸性水解实现脱保护,分别以66%和67%的产率获得目标四氢噻吩砜衍生胺顺式-8c和反式-8c。
相比之下,用间氯过氧苯甲酸处理13a未取得成功,仅产生痕量目标氨基酸衍生物13c。通过在催化量钨酸钠(Na₂WO₄)[45,46]存在下,13a与30%过氧化氢水溶液(w/w)反应实现了选择性硫氧化。随后的脱保护步骤通过两步反应以46%的产率获得氨基酸盐酸盐11c(5g规模)。
2.2 酸碱性质
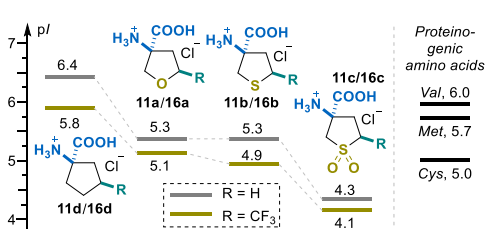
通过标准酸碱滴定法测定了化合物顺式-8a-d、反式-8a-c、11a-c、环亮氨酸(16d)及其三氟甲基取代衍生物11d(二元解离)的pKa值(图2和图3)[47]。为深入了解特定片段对可电离基团酸度的影响,将已报道的非氟代杂环胺17a-c[48]和环戊胺17d[49]的相关数据纳入分析。
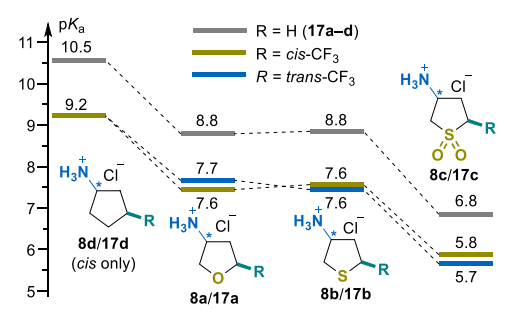
正如预期的那样,盐酸盐8a-d的酸度主要受电子因素影响,四氢噻吩砜衍生物8c的pKa值最低(顺式和反式异构体分别为5.7和5.8)(图2)。硫/亚砜(S/SO₂)取代的净效应与在环戊烷核心中引入单个杂原子的效应相当(8c/9b和9b/9d对的ΔpKa值分别为1.8和~1.6)。观察到的差异与甲氧基(OCH₃)、甲硫基(SCH₃)和甲磺酰基(SO₂CH₃)的Swain–Lupton场参数F(分别为0.29、0.23和0.53)[50]相关,这证实了可电离铵部分的pKa值主要受诱导效应控制。
在环戊胺(17d,R=H)中引入三氟甲基片段,使氨基的碱性降低了1.3个pKa单位(图2)[51]。同时,杂环胺8a-c中三氟甲基的pKa增量在1.0-1.3个pKa单位范围内,表明杂原子的电子效应仅在一定程度上具有加和性。在所有情况下,顺式和反式异构体的pKa值差异较小(ΔpKa不超过0.1个单位)。
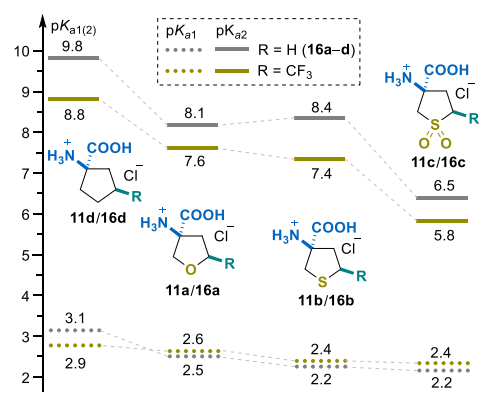
氨基酸盐酸盐11a-d的pKa数据遵循化合物8a-d系列的变化趋势:环烷烃>>四氢呋喃/四氢噻吩>四氢噻吩砜(图3)。对实验测定的pKa值的深入分析表明,第一电离常数(pKa1)在氨基酸系列中的变异性有限,范围为1.0个pKa单位(四氢噻吩砜16c为2.2,非氟代环戊烷衍生物16d为3.1)。此外,含氟和非含氟化合物的比较表明,三氟甲基化对羧基(CO₂H)片段酸度的提升有限(0.1-0.2个pKa单位)。相反,引入三氟甲基后pKa2显著降低,尽管13a-d的ΔpKa2值略低于胺盐酸盐的ΔpKa 值(13a-d为0.6-1.1个单位,8a-d为1.0-1.3个单位)。同样,四氢噻吩砜11c的ΔpKa2值最小(0.6个pKa单位),这可能与两个强电子受体的电子效应部分抵消有关。
杂原子效应呈现出类似趋势。因此,对于非氟代和三氟甲基取代衍生物,用亚砜(SO₂)取代硫(S)分别使pKa值降低了2.0和1.5个单位。同时,在环戊烷骨架中引入杂原子(硫或氧),对于非氟代和三氟甲基取代衍生物,分别使pKa值降低了1.4-1.7和1.3-1.5个单位。
等电点(pI)比较表明,所有取代环亮氨酸类似物的pI均如预期般降低,CH₂→O/S和O/S→SO₂取代的ΔpI均约为1个单位(图4)。三氟甲基化的影响虽不显著但仍值得关注:引入三氟甲基部分导致pI降低0.2-0.6个单位。这种效应在环戊烷衍生物中最为明显(16d/11d对的ΔpI=0.6),而本身已含有吸电子基团的四氢噻吩砜衍生物11c的ΔpI仅为0.2个单位。在蛋白原性天然氨基酸中,三氟甲基取代的四氢噻吩和四氢呋喃衍生物11a和11b与半胱氨酸最为相似(pI分别为4.8、5.1和5.0)。相反,四氢噻吩砜类似物11c表现出明显更高的酸性,在蛋白原性氨基酸中没有直接对应的类似物。
2.3 亲脂性
采用标准摇瓶法结合高效液相色谱(HPLC)定量分析,测定了苯甲酰胺顺式-18a-c和反式-18a-c的LogP值(详见支持信息)[52]。与由杂原子电负性控制的单调酸度趋势不同,亲脂性遵循独特的规律,主要受取代基极化率、空间位阻和氢键能力的影响。
在研究系列中,四氢噻吩衍生物表现出最高的LogP值(顺式-18a和反式-18a分别为2.82和2.71),而其砜类似物18c的亲脂性显著更低(分别为1.57和1.54)(图5)。这种显著差异反映了硫的固有性质:硫原子比氧原子更大、电负性更低,且与亚砜(SO₂)片段相比,形成氢键的能力显著较弱。四氢呋喃衍生物表现出中等的LogP值,比母体N-苯甲酰基环戊胺(18d)低0.32-0.47个单位。取代基的相对位置对亲脂性的影响较小,最大ΔLogP为0.17个单位。
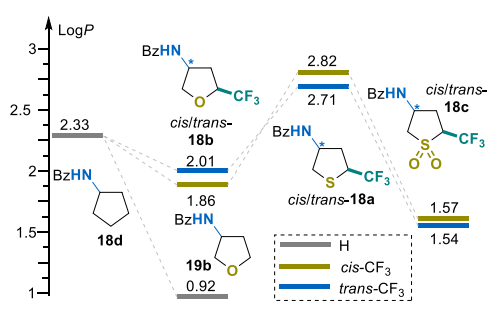
等电点(pI)在CH₂→O/S→SO₂取代或三氟甲基化时均呈现系统性降低。LogP测定结果表明,所研究系列中杂原子的亲脂性趋势为:S>CH₂>O>SO₂。尽管三氟甲基化如预期般使化合物性质向酸性增强和亲水性提高的方向转变,但并未改变由环骨架中(杂)原子性质(CH₂/S/O/SO₂)定义的固有趋势。换句话说,观察到的pKa 和 LogP变化主要依赖于杂环骨架。在整个系列中,顺式和反式立体异构体仅表现出微小差异。
我们认为,所提出的化学型是药物研发项目中极具前景的构建块,不久将在药物化学和拟肽化合物设计中得到应用。特别是,合成的含氟杂环氨基酸可被视为非蛋白原性氨基酸环亮氨酸的类似物,具有多种生物医学应用。
3、结论
本研究开发了一种可规模化且用途广泛的合成方法,以商业可得的起始原料(即(硫代)乙醇酸酯和4,4,4-三氟巴豆酸酯)合成含氟和非含氟四氢呋喃和四氢噻吩衍生构建块。我们对该方法(此前仅报道用于三氟甲基取代四氢噻吩)的改进包括在第一步反应中更换碱,这简化了分离流程(即避免水相后处理)并提高了总产率(三步反应产率从66%提升至77%)。此外,该方法首次扩展至四氢呋喃衍生物的合成。优化后的方案能够以十克级规模高效制备三氟甲基取代酮类化合物及其非对映纯顺式和反式异构体醇、胺和氨基酸衍生物,以及四氢噻吩砜类似物。
对所提出化学型的理化性质进行了表征。pKa值分析表明,杂原子(尤其是亚砜基团)具有强诱导效应,三氟甲基取代基可进一步调节酸度。LogP测定结果揭示了杂原子相关的亲脂性趋势(S>CH₂>O>SO₂),且立体异构体对理化性质的影响较小。这些结果为含氟杂环砌块在药物设计中的合理应用提供了重要的理化数据支持。
对于本文中提到的中间体或砌块,Enamine都备有现货库存:
阅读原文了解更多详情