1、摘要
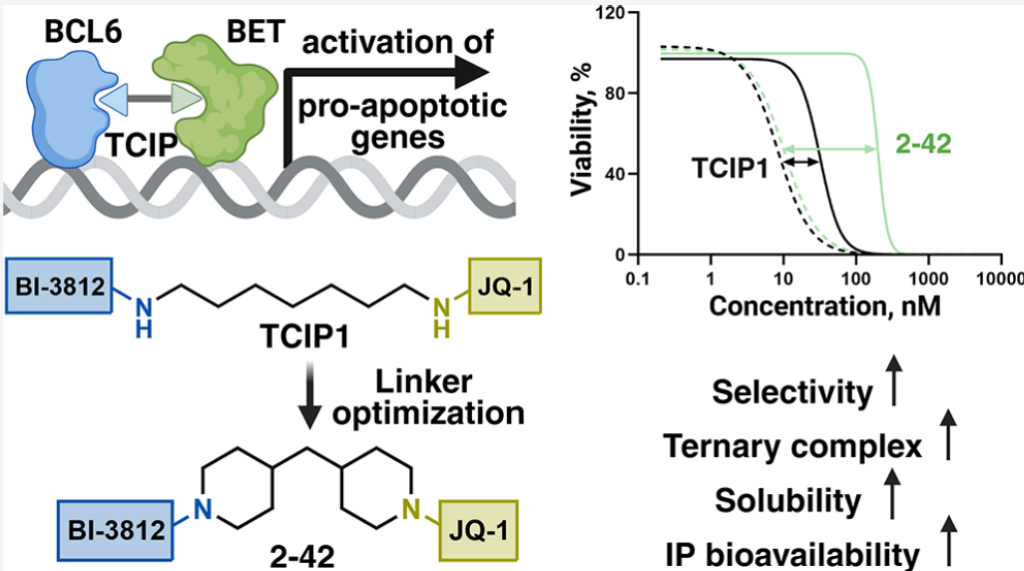
B细胞淋巴瘤6(BCL6)是一种转录阻遏蛋白,与弥漫性大B细胞淋巴瘤及其他恶性肿瘤密切相关。传统BCL6抑制剂和降解剂依赖于功能丧失机制,可能因通路抑制不完全而受限。转录邻近化学诱导剂(TCIPs)提供了一种功能获得性替代策略,通过将转录共激活因子重定向至BCL6结合的基因组位点,从而重新激活促凋亡基因的表达。本文系统描述了通过连接子工程化进行BCL6 TCIPs的药物化学优化。以JQ1和BI-3812为基础构建了包含66个杂双功能类似物的聚焦化合物库,评估了三元复合物形成能力、细胞活性和选择性。连接子刚性化及环状元素的引入显著提高了细胞选择性,增强了溶解度,并增加了小鼠体内的血浆暴露量。计算分析、竞争实验和RNA测序结果表明,优化类似物的作用由三元复合物形成所驱动。综上,这些发现确立了连接子架构作为TCIP性能关键决定因素的地位。
2、引言
转录阻遏蛋白B细胞淋巴瘤6(BCL6)在沉默凋亡相关基因中发挥关键作用,对生发中心——产生持久体液免疫和B细胞增殖所必需的微解剖结构——的形成至关重要。然而,BCL6的持续表达及下调障碍可导致细胞死亡相关基因的持续阻遏,进而引起B细胞不受控增殖和肿瘤发生。¹
弥漫性大B细胞淋巴瘤(DLBCL)是非霍奇金淋巴瘤最常见的亚型,也是研究最为广泛和深入的BCL6驱动型癌症。据估计,2025年仅美国就将诊断超过20,000例新病例,预计到2032年将超过30,000例。²,³ 标准DLBCL治疗方案通常为化疗联合免疫治疗,最典型的是R-CHOP方案(利妥昔单抗、环磷酰胺、多柔比星、长春新碱和泼尼松),此外还包括放射治疗、抗体靶向治疗、CAR-T细胞治疗和干细胞移植。尽管约60-70%的患者可达缓解,但30-40%的患者将在完成治疗后两年内复发,目前尚无获批的替代疗法。⁴
除DLBCL外,BCL6上调还在多种其他恶性肿瘤中被观察到,包括滤泡性淋巴瘤、B急性淋巴细胞白血病、混合谱系白血病、伊马替尼耐药慢性髓系白血病、急性髓系白血病、非小细胞肺癌和三阴性乳腺癌。¹ BCL6的广泛参与使其成为极具吸引力的治疗干预靶点。
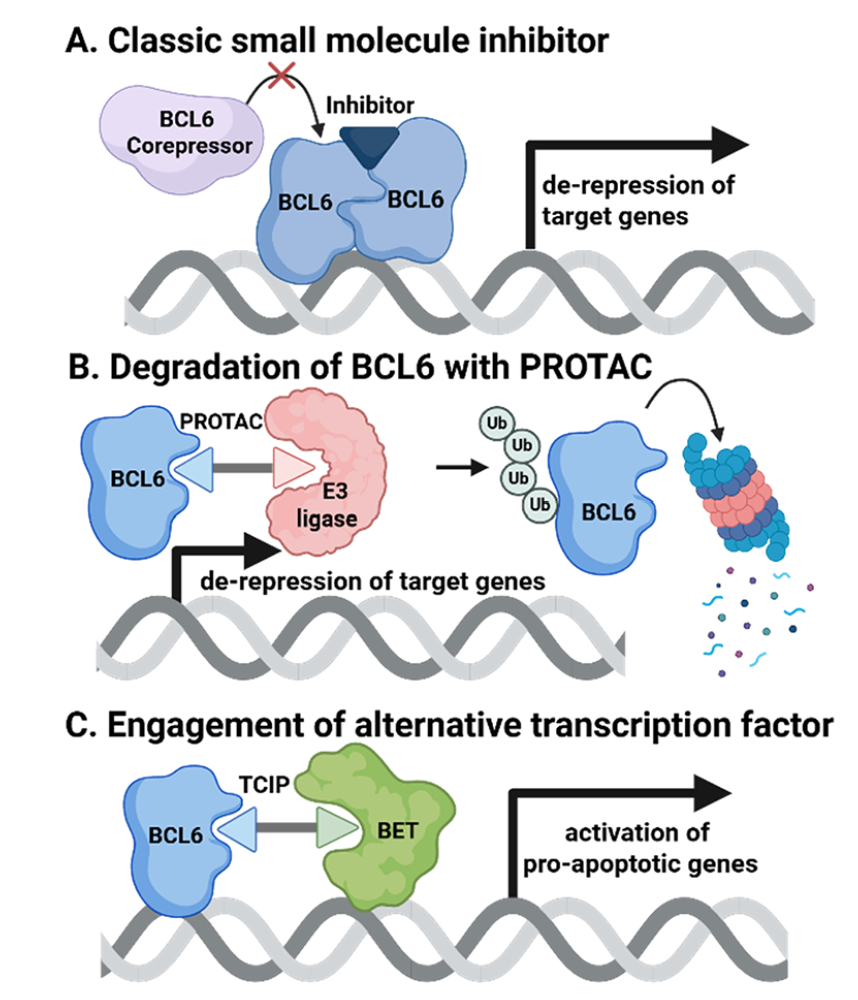
近年来,众多学术机构、生物技术公司和制药企业加大了BCL6抑制剂和降解剂的开发力度(图1A,B)。⁵ BCL6抑制剂(如BI-3812⁶或OICR12694⁷)与BCL6的BTB结构域结合,干扰共阻遏因子的结合。BCL6 BTB结构域抑制剂的安全性已在携带突变BTB结构域(无法结合共阻遏因子)的小鼠中得到验证,这些小鼠保持健康且未出现炎症综合征迹象。⁸ 然而,尽管现有BCL6抑制剂具有前景良好的临床前活性,但无一进入临床试验。原因可能在于,仅抑制共阻遏因子结合不足以诱导持续的理想治疗效果,需探索不同的靶向模式。值得注意的是,目前有两项临床试验正在评估BCL6蛋白水解靶向嵌合体(PROTACs),分别由Arvinas⁹和Bristol Myers Squibb¹⁰开发。这些及近期报道的PROTACs¹¹⁻¹⁴通过清除细胞中的BCL6蛋白发挥作用,提供了优于单纯抑制的治疗效果(图1B)。然而,这些药物属于"功能丧失"策略,可能需要高胞内药物浓度,潜在导致脱靶效应或疗效有限,尤其在大体积或血管化不良的肿瘤中。AstraZeneca 2018年的一项研究¹⁵ 佐证了这一局限性:BCL6降解剂虽实现了显著降解和高胞内PROTAC浓度,却未能引发预期表型效应——可能归因于残留的BCL6活性。此外,虽然BTB抑制已被证明安全,但BCL6敲除小鼠中BCL6的完全丢失会导致致死性炎症表型。⁷
为应对抑制剂和PROTACs面临的挑战,一种"功能获得性"策略应运而生——转录邻近化学诱导剂(TCIPs),由Crabtree和Gray实验室首创。¹⁶,¹⁷ 该创新策略建立在两个蛋白质间邻近诱导的概念之上,¹⁸,¹⁹ 作为一种前景广阔的替代方案,值得生物技术界给予更多关注。²⁰⁻²² 简言之,TCIPs利用转录因子的DNA结合特异性,通过将转录激活因子系拴至BCL6,将这些共激活因子重定向至BCL6结合的启动子,诱导通常被沉默的促凋亡基因表达(图1C)。与传统功能丧失疗法不同,TCIPs采用功能获得性机制,利用癌细胞自身的转录机器触发细胞毒性基因表达,仅需部分靶标被占据即可起效,无需完全抑制或降解,从而可能避免抑制必需蛋白时常见的靶向毒性。
TCIPs的开发前景广阔,但这些双功能分子超出了类药五规则范围,需进行大量优化以实现生物利用度。原始出版物中报道的先导化合物TCIP1所含的线性庚基(C7)连接子存在若干缺陷,预期代谢稳定性和溶解度较差。近期研究表明,连接子优化是药代动力学优化的主要驱动因素之一,尤其是当两个蛋白的结合配体已高度优化时。²³ 例如,临床阶段PROTACs通常采用含单个叔胺基团的连接子,如哌啶、哌嗪或氮丙啶。此外,使用环状结构作为构象限制是口服生物利用度PROTACs中反复出现的策略,大多数实例的连接子含至少两个环,部分甚至含三个。许多设计还在环间引入亚甲基间隔基,以平衡柔性与空间排列。²⁴
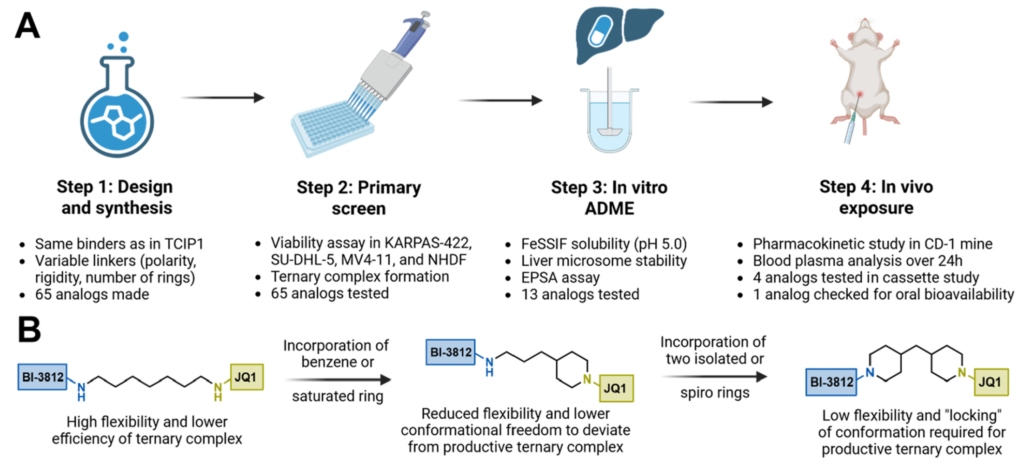
Arvinas和AstraZeneca的近期研究强调了减少未满足或溶剂暴露的氢键供体(eHBDs)数量对增强口服吸收的重要性。²⁵,²⁶ 基于上述认识,我们旨在设计更优化的连接子来连接JQ1和BI-3812构建模块,双重目标是改善三元复合物形成效率并增强靶向细胞毒性,遵循图2的工作流程。此外,我们希望探究连接子设计作为实现物理化学和药代动力学特性主要驱动因素的潜力,使之与已验证的生物利用度双功能化学型(如ARV-110、²⁷ KT-474、²⁸ NX-5948²⁹)相当。
3、结果与讨论
为合成TCIP1的类似物,我们使用了Enamine的连接子集合,³⁰ 包含近8000个现货可用构建块,涵盖多种长度、额外杂原子、构象限制元素和正交官能团组合。基于JQ1和BI-3812的化学性质(方案1A),选择了单N-Boc保护二胺连接子。在此类650个可用连接子中,按以下标准筛选了65个代表性连接子1:(i)连接子长度为6-8个原子(不含官能团);(ii)结构多样性,涵盖无环、醚、叔胺、苯环、环己烷、一个或两个饱和杂环胺环(即氮丙啶、哌啶、吡咯烷和哌嗪)或螺环片段;(iii)可及性与成本效率(方案1B和图S1)。含(保护的)伯胺和仲胺官能团的化合物也纳入选择。先前作者用作TCIP1¹¹ 的1,7-庚叉连接子亦作为参照纳入。
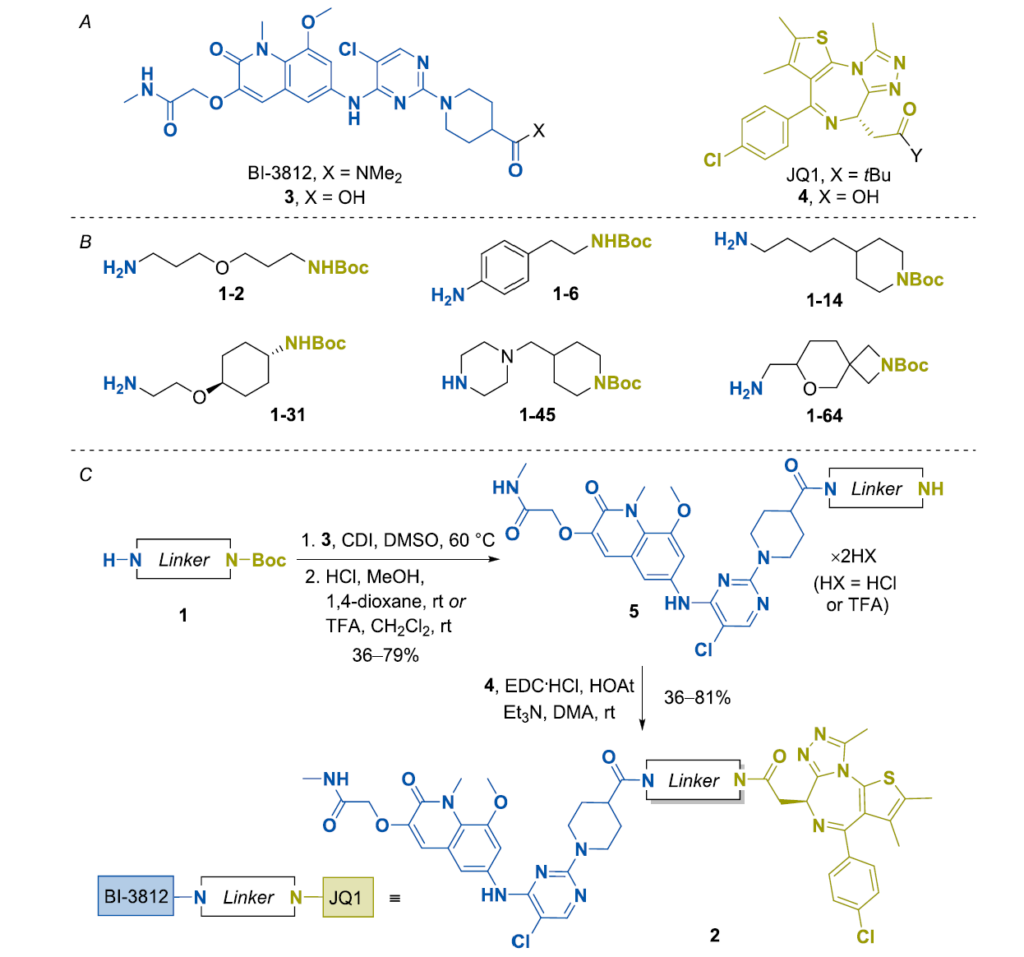
目标双功能分子2的合成从羧酸3和4——分别为BI-3812和JQ1的衍生物——出发(方案1C)。为促进化合物1和3的反应,以CDI为COOH基团活化剂、DMSO为溶剂,在60°C下进行。在早期实验中,所得粗品酰胺以MeOH/1,4-二氧六环中的HCl进行脱保护。但部分情况下,反应因3的甲酯副产物形成而复杂化,该副产物难以与中间体分离。因此,后续实验改用CH₂Cl₂中的TFA去除N-Boc保护。所得盐5在EDC和HOAt存在下与羧酸4反应。经反相HPLC纯化,目标产物2以13-55%的总产率获得
工作流程的下一步是使用BCL6阳性细胞系(SU-DHL-5和KARPAS-422)以及BCL6阴性细胞系MV4-11对所有类似物进行细胞活力测定。选择MV4-11作为BCL6阴性代表模型,原因在于与其他两种细胞系类似,它属于造血系统肿瘤、悬浮生长,且对BET抑制高度敏感。优先选择在MV4-11中EC₅₀与SU-DHL-5和KARPAS-422中EC₅₀相比选择指数最高的化合物,以鉴定其活性主要由BCL6和BET蛋白间三元复合物形成驱动、而非BCL6非依赖性BET抑制驱动的候选化合物。
首先评估了具有高柔性和最小复杂度的连接子的构效关系——完全线性或仅含单个哌啶、哌嗪或氮丙啶环(表1)。在连接子中心插入氧原子(2-1)使BCL6表达细胞系中的活性降低3-6倍;在BI-3812侧缩短连接子(2-2)则降低了SU-DHL-5和MV4-11中的活性。在连接子中心添加叔胺(2-3)进一步降低效力。
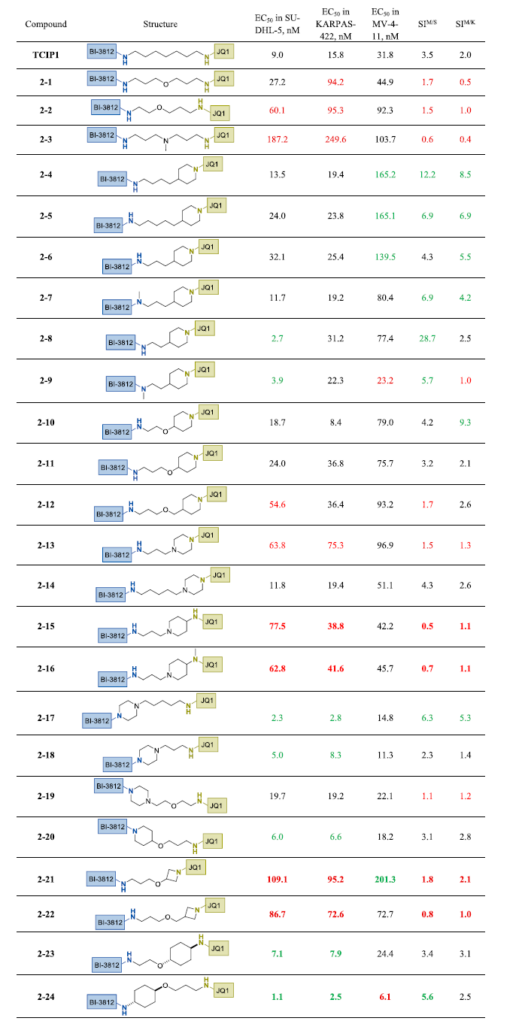
最小刚性化策略——如在JQ1侧引入哌啶(2-4)——降低了MV4-11毒性并提高了选择性(相比TCIP1提高>3倍)。细微的连接子长度调整(2-5、2-6)在效力中等降低的情况下仍保持了选择性。有趣的是,BI-3812酰胺的N-甲基化(2-7)使SU-DHL-5效力增强约3倍,同时略微降低了MV4-11的EC₅₀。进一步缩短含哌啶连接子(2-8)使选择指数(SI)最大化,SI由MV4-11中EC₅₀除以SU-DHL-5(SIM/S)或KARPAS-422(SIM/K)中EC₅₀计算。氧原子引入的效果呈上下文依赖性:短连接子(2-6→2-10)中活性得以保持,而长连接子(2-4、2-5)中效力和选择性降低(2-11、2-12)。哌啶到哌嗪的替换结果不一:2-13活性较差,2-14保持效力但选择性下降。将哌嗪重新连接至BI-3812(2-17)提高了所有细胞系的活性并维持选择性;缩短该连接子(2-18)降低选择性,而氧原子引入(2-19)则完全消除选择性。含氮丙啶连接子(2-21、2-22)降低了活性和选择性。引入反式-1,4-环己基氨基醚片段对效力的影响取决于位置:靠近BI-3812放置(2-24)产生了该系列中活性最高的化合物,在SU-DHL-5和KARPAS-422中的活性比TCIP1高6-9倍,且改善了对MV4-11的选择性。
下一组连接子含有1,4-或1,3-二取代苯环(表2)。BI-3812旁含苯环的2-25表现出与原始TCIP1相当的活性和选择性。JQ1侧酰胺的N-甲基化(2-26)导致活性降低,选择性保持相似。2-25的连接子含额外碳或氧的类似物(2-27和2-28)显示效力改善,但无选择性收益。
将苯环移至连接子中心(2-29和2-30)对活性影响甚微。相反,将苯环定位至更靠近JQ1部分显著增强了选择性。较短和较长的连接子类似物2-31和2-32均显示选择性改善,主要归因于MV4-11细胞毒性降低。在2-30中用氮丙啶替换乙二胺片段对活性影响甚微(2-33)。然而,对2-25施加类似修饰所得2-34在BCL6表达细胞中显示效力和选择性改善。通过旋转氮丙啶或以哌嗪替换,2-35和2-36获得了进一步改善。值得注意的是,这些情况下选择性增强同样由MV4-11细胞毒性降低驱动,尽管苯环更靠近BI-3812侧。以亚甲基哌啶替换2-27中的柔性连接子显著降低总体活性,但将MV4-11中EC₅₀值移至微摩尔范围,从而增强了对BCL6表达细胞的选择性。所得化合物2-37为本系列中选择性最高的化合物之一;然而其效力——尤其是KARPAS-422细胞中——对于药代动力学受限的双功能分子可能不足。有趣的是,将芳基连接子上的连接点移至间位(2-38)仅略微降低选择性。芳基连接子系列的最后一个类似物——将哌啶连接至BI-3812(2-39)——表现出适度的活性和选择性。
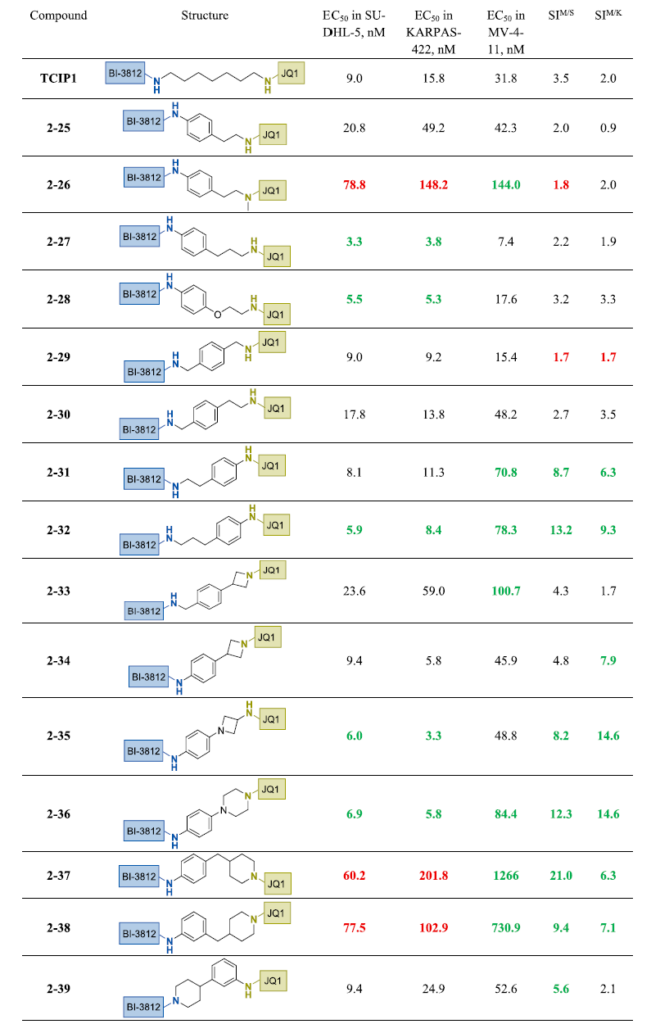
如表1所示,即使是通过添加哌啶环实现的轻微刚性化,也改善了最终双功能分子对BCL6表达细胞系的选择性。基于此观察,我们接下来探讨了添加第二个饱和环的效果(表3)。由亚甲基分隔的哌嗪和哌啶组合产生了高效且高选择性的类似物2-40和2-41。当其中一个哌嗪环替换为哌啶时,所得类似物2-42在保持高选择指数的同时降低了对MV4-11细胞的毒性。在连接子中引入额外氧原子(2-43)对效力或选择性无显著影响。在2-41的连接子中添加额外亚甲基略微降低效力;但2-44仍保持良好选择性。将2-40中的哌嗪替换为4-氨基哌啶得类似物2-45,活性相似但对MV4-11细胞毒性增加,选择指数降低。有趣的是,从4-氨基哌啶转为氮丙啶降低了2-46在所有三种细胞系中的毒性——这一趋势在化合物2-21、2-22和2-33中亦有观察到。将哌啶替换为哌嗪略微改善效力,但2-47选择性仍较低。在2-46的连接子中引入氧原子改善了2-48的效力(尤其是KARPAS-422中),并降低了MV4-11细胞毒性,从而显著提高了该细胞系的选择性。当3-氨基氮丙啶代替4-氨基哌啶用于2-45时,2-49及其低级同系物2-50在所有三种细胞系中保持低纳摩尔活性,选择性较差。以哌嗪替换3-氨基氮丙啶(2-51)略微增加MV4-11细胞中EC₅₀,部分恢复选择性。含两个孤立饱和环的最具前景化合物——2-52、2-53和2-54——基于氮丙啶、吡咯烷或哌啶与反式-1,4-环己基氨基醚部分的组合。值得注意的是,将哌啶环上醚键连接位置从C-4移至C-3显著降低了2-55在MV4-11细胞中的EC₅₀,从而降低选择性。
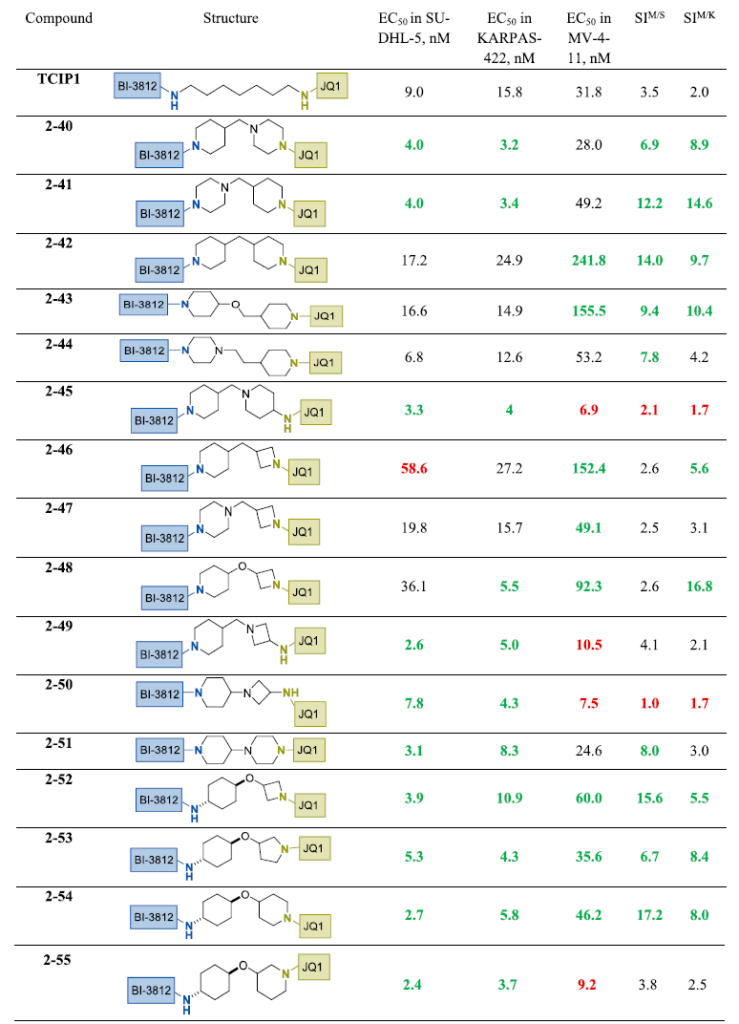
近年来,螺环框架因提高分子三维性、降低总极性表面积和改善药代动力学特性的能力而备受关注。³¹ 为研究螺环连接子对TCIP1类似物效力和选择性的影响,我们在设计中引入了若干代表性螺环基序(表4)。
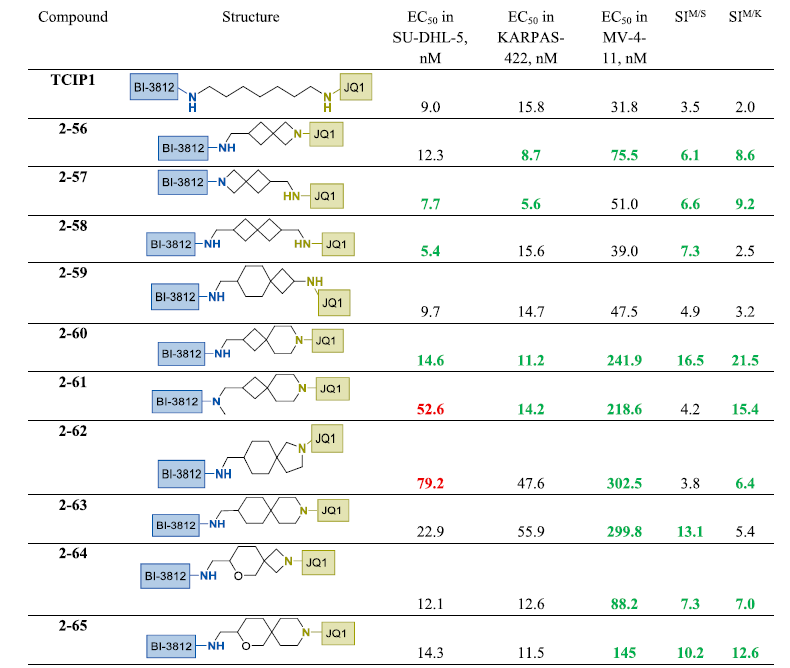
在连接子中引入2-氮杂螺[3.3]庚烷片段并连接至JQ1或BI-3812结合部分,适度增强了KARPAS-422细胞效力并降低了MV4-11细胞毒性,从而改善了总体选择性,如化合物2-56和2-57所示。相反,在连接子中间引入螺[3.3]庚烷单元仅使2-58在SU-DHL-5细胞中效力和选择性略有提升,对KARPAS-422和MV4-11的影响可忽略不计。含螺[3.5]壬烷核心的化合物2-59表现出与TCIP1相当的生物学特征。值得注意的是,以直接连接至JQ1部分的7-氮杂螺[3.5]壬烷替换该基序,所得2-60成为本研究中鉴定的最具选择性的TCIP1类似物之一。有趣的是,BI-3812侧酰胺的N-甲基化(2-61)显著降低SU-DHL-5细胞活性,同时在KARPAS-422和MV4-11中保留效力。进一步探索螺环骨架发现,含2-氮杂螺[4.5]癸烷单元的2-62在所有测试细胞系中活性降低。相比之下,密切相关的3-氮杂螺[5.5]十一烷类似物2-63在SU-DHL-5中表现改善,尽管KARPAS-422中效力仍为中等(约50 nM)。最后,含6-氧杂-2-氮杂螺[3.5]壬烷(2-64)和2-氧杂-9-氮杂螺[5.5]十一烷(2-65)环系的类似物在BCL6表达细胞系中维持了与TCIP1相当的活性,同时在MV4-11细胞中表现出3-4倍更低的毒性,选择性得以提高。
总体而言,本研究评估的连接子变体中约50%在SU-DHL-5、KARPAS-422和MV4-11细胞系的细胞活力测定中表现出相比TCIP1增强的效力或选择性。KARPAS-422和SU-DHL-5间观察到的效力呈良好相关性(图S3A)。为优先筛选化合物进行进一步研究,我们整合了正常人皮肤成纤维细胞(NHDFs)活力数据和NanoBiT三元复合物形成数据(详见表S1和图S2)。部分化合物(如2-54、2-52和2-65)对BCL6表达细胞相对于MV4-11显示极好的选择性,但在成纤维细胞活力测定中EC₅₀值较低,因此未进入工作流程的下一步。
为表征三元复合物形成,我们采用NanoBiT系统评估了三个参数:TCmax(归一化至TCIP1的最大观察信号)、EC₅₀(三元复合物形成达到TCmax 50%时的浓度)和AUC(归一化至TCIP1的剂量-响应曲线下面积)。将各参数对选择性作图(图S3B-D)显示,EC₅₀对选择性三元复合物形成的预测性最弱,而TCmax和AUC表现出最强关联性。
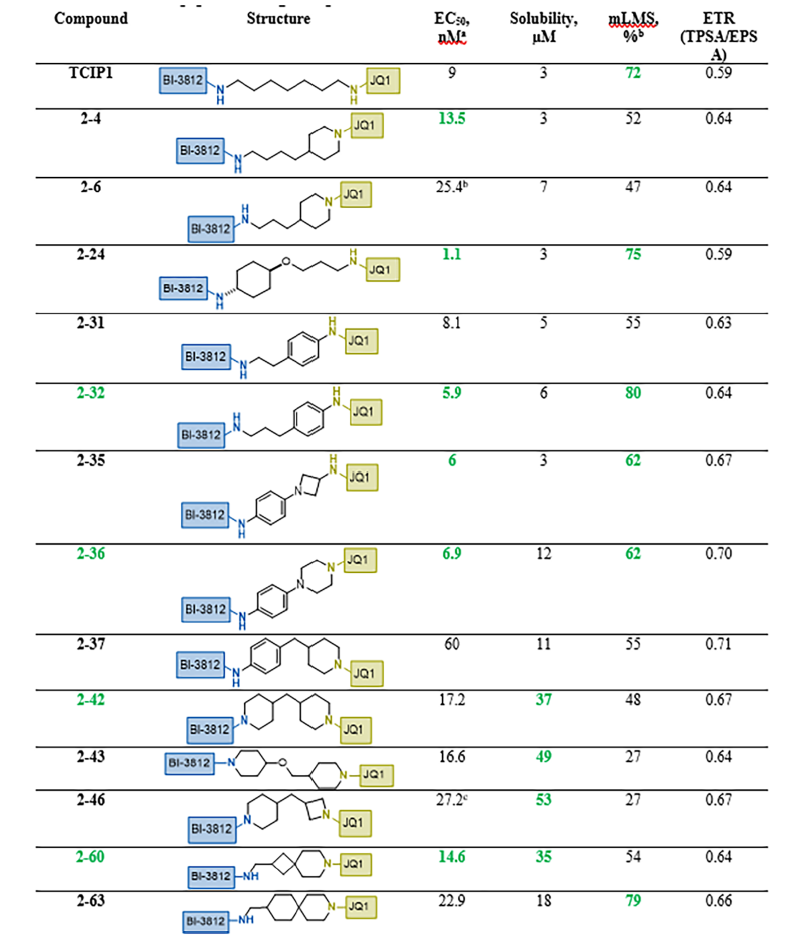
基于综合分析,筛选出13个最具效力和选择性的化合物进行物理化学表征(表5),包括FeSSIF溶解度、小鼠肝微粒体稳定性(mLMS)和实验极性表面积(EPSA)测定。其中约一半化合物的溶解度相比TCIP1有至少3倍改善。代谢稳定性方面,大多数化合物表现出良好的小鼠肝微粒体稳定性,其中2-24、2-32和2-63显示出该组中最高的稳定性值(75-80%)。作为吸收预测指标,我们计算了近期引入的"极性降低"描述符ETR——定义为拓扑极性表面积(TPSA)与实验极性表面积(EPSA)之比。³² 根据DeGoey等人的观点,TPSA > 140的分子需ETR低于0.8方能在啮齿动物中实现肠道吸收。表5所列化合物的ETR值在0.59至0.70之间,表明测试分子具有高极性降低能力和"变色龙性"。正如预期,柔性较高、刚性最低的连接子显示约0.6的ETR值(TCIP1和2-24),而刚性化双环化合物(2-37和2-36)的ETR值接近0.7。
基于表5结果,选择四个先导候选化合物进行CD-1小鼠药代动力学(PK)盒式给药评估。包括苯连接类似物(2-32)、含苯基哌嗪双功能化合物(2-36)、含两个孤立饱和环类似物(2-42)和螺环连接子类似物(2-60),以TCIP1为参照。四个化合物在BCL6阳性细胞中均表现出改善的选择性和三元复合物形成效率(图3)。
血浆浓度-时间曲线(图4A)显示,2-42——连接子中含两个孤立哌啶环的TCIP1类似物——表现出最高的血浆暴露量(AUC,图4B)。给药后105分钟达到最大血浆浓度(Cmax)2040 ng/mL(约2 μM),血浆半衰期(T₁/₂)约5-6小时。连接子含一个芳香环的类似物(2-32)血浆暴露量低2倍,但半衰期更长(约10小时)。相比之下,螺环类似物2-60和含苯基哌嗪类似物2-36均显示较差的血浆暴露量。对照化合物TCIP1在测试条件下溶解度不足,被排除在分析之外。此外,使用10%血浆进行24小时透析的血浆蛋白结合测定以确保达到平衡,所得结合值随后外推至100%血浆条件。2-36在测试条件下似不稳定,解释了其较差的暴露量。2-60和2-32的蛋白结合率分别为99.6%和99.4%,TCIP1和2-42则非常相似——分别为99%和99.1%。考虑血浆蛋白结合,2-42在5 mg/kg静脉给药后的未结合血浆浓度约为20 ng/mL(约20 nM)。
为评估2-42的口服生物利用度,在静脉和口服给药后测定血浆浓度(图4C-D)。口服给药后血浆暴露量远低于静脉或腹腔给药(2-42腹腔给药数据来自盒式研究)。口服暴露量低最可能归因于2-42渗透性较差,如MDCK-MDR1测定结果所示(图4B)。
为探究2-42相对于TCIP1形成更高效三元复合物的基础,我们对两种三元复合物进行了分子动力学(MD)模拟。2-42和TCIP1共享靶向BCL6和BRD4的相同弹头部分,仅连接子架构不同:TCIP1含柔性庚亚甲基连接子,2-42引入构象受限的双哌啶连接子。尽管端到端长度相当,两种连接子具有根本不同的构象性质。MD模拟揭示,在三元复合物内,TCIP1连接子的结合构象以沿烷基链多个gauche扭转为特征,偏离能量最优的全反式几何,在结合配体中引入扭转应变。相比之下,2-42的双哌啶连接子在未结合态下构象柔性大幅降低,六元环的几何约束促进了对生物活性构象的预组织,从而降低三元复合物组装的熵代价(图5A)。
使用Freeform(OpenEye)进行的独立扭转分析证实了上述观察。2-42的结合构象被确定为其构象能量表面的全局最小值(Erel = 0.00 kcal/mol,conf_dG = 1.05 kcal/mol,population = 17.08%),而TCIP1的结合构象位于其全局最小值之上2.25 kcal/mol,仅在8.61%的可及构象体中存在(conf_dG = 1.45 kcal/mol)。该约2 kcal/mol的结合态应变能惩罚,加之生物活性构象体较低的平衡群体,与TCIP1降低的三元复合物效率一致,支持了连接子预组织是本系列活性关键决定因素的结论。
为探究该基于应变的原理的普适性,我们将分析扩展至第二对匹配化合物2-54和2-55。与2-42和TCIP1的对比不同,2-54和2-55的应变曲线几乎不可区分:两者在结合态均采用各自全局最小构象(两者的Erel = 0.00 kcal/mol),conf_dG值分别为1.45和1.24 kcal/mol,生物活性构象体群体分别为8.61%和12.36%。虽这些值表明2-55应变略低,但实验数据显示2-54是活性更高的化合物。因此,配体应变和构象熵不太可能是该对化合物活性的主要决定因素,暗示其他因素亦贡献于观察到的差异。
使用AMBER³³,³⁴中MM/GBSA方法计算所有四个三元复合物的结合自由能,按与实验活性一致的方向对匹配化合物排序。2-42表现出最有利的结合能-159.6 ± 1.0 kcal/mol(均值 ± S.E.),TCIP1为-150.1 ± 0.7 kcal/mol,差值约9.5 kcal/mol,印证了上述基于应变和预组织的原理。对于第二对匹配化合物,2-54预测比2-55形成更稳定的三元复合物(-154.7 ± 0.7 vs -147.8 ± 0.8 kcal/mol),再现了实验观察的活性排序,尽管两化合物间并无有意义的应变差异。
综上,Freeform应变分析、MD模拟和MM/GBSA分析表明,连接子预组织决定了2-42和TCIP1间的活性差距,而2-54和2-55间的差异则由三元复合物内相互作用能主导。这些互补贡献被MM/GBSA结合能所捕获,为未来连接子设计提供了机制框架。
为评估二元相互作用的贡献,我们使用预形成的三元复合物进行了竞争实验。BET溴结构域抑制剂JQ1以相似EC₅₀值(22-43 nM)置换TCIP1、2-42、2-54和2-55形成的三元复合物,尽管2-54对竞争的抵抗力略强(图5B)。BCL6抑制剂BI-3812以相似EC₅₀值(1.9-3.8 nM)置换这些三元复合物,表明三元复合物内测试化合物的二元亲和力相当,尽管2-42形成的复合物需稍高竞争剂浓度方可解离(图5C)。我们还使用SmBiT-BCL6和LgBiT-HaloTag靶标参与测定(BRD4-BCL6三元复合物非依赖方法)进行了BCL6亲和力评估,观察到EC₅₀值在4至22 nM之间(图5D),其中TCIP1和2-54分别以最低和最高亲和力结合BCL6。这些结果进一步支持了2-42和2-54优于TCIP1和2-55的优异性能由多种因素驱动的结论,包括二元亲和力、构象应变和三元复合物内相互作用能。
为进一步验证2-42在BCL6阳性细胞中的抗增殖活性由三元复合物形成而非二元靶标参与驱动,我们进行了细胞竞争实验。以固定浓度2-42处理SU-DHL-5细胞,并共给予递增浓度的BCL6抑制剂BI-3812(该化合物在此细胞系中浓度低于1 μM时无活性)。BI-3812以剂量依赖方式恢复细胞活力,EC₅₀为27 nM,证实BCL6处三元复合物形成是2-42细胞活性所必需的,且仅二元BRD4参与不足以驱动观察到的抗增殖效应(图6A)。最后,对以100 nM 2-42处理20小时的样品进行RNA测序分析,显示MYC受抑、BCL6靶基因上调及促凋亡基因激活,与Crabtree和Gray实验室对TCIP1的观察一致(图6B)。
4、结论
本研究系统探索了TCIP类似物——旨在诱导BCL6和BRD4间三元复合物形成的双功能分子——的连接子空间。从涵盖无环、芳基、饱和和螺环架构的65个单N-Boc保护二胺构建块出发,我们合成并评估了TCIP类似物库,鉴定了控制BCL6表达淋巴瘤细胞系效力和选择性的关键结构决定因素。
四类连接子的构效关系揭示了清晰且一致的设计原则:(1)在柔性连接子中引入杂原子同时降低活性和选择性;(2)引入饱和环或苯环通常改善选择性并增强三元复合物形成;(3)苯环与饱和环组合、两个饱和环或螺环的引入进一步强化选择性和三元复合物形成;(4)在更刚性连接子中包含杂原子不影响活性或三元复合物形成。整个系列中,选择性改善始终由BCL6阴性MV4-11细胞系中脱靶细胞毒性降低驱动,而非BCL6表达细胞内在效力提升。
基于改善的活性、选择性、溶解度及良好的肝微粒体稳定性,四个先导化合物(2-32、2-36、2-42和2-60)被推进至CD-1小鼠药代动力学评估。含构象受限双哌啶连接子的化合物2-42成为药代动力学最优候选。分子动力学和竞争实验表明,二元亲和力、构象应变和三元复合物内相互作用能等多种因素共同贡献了2-42及其他TCIP类似物的活性。BI-3812细胞挽救实验和RNA测序分析进一步验证了2-42在BCL6阳性细胞中的抗增殖活性由靶向三元复合物形成驱动,再现了先前报道的TCIP1转录特征。
尽管具备上述有利特性,渗透性仍是关键限制因素。化合物2-42在MDCK-MDR1测定中表观渗透性较低(Papp A-B = 0.6 × 10⁻⁶ cm/s),虽FeSSIF中溶解度(37 μM)可接受,但在啮齿动物中5 mg/kg口服给药后生物利用度较低,尽管腹腔暴露量高且代谢稳定性优良(清除率1.8 mL/min/kg)。这些结果表明,连接子优化虽可大幅改善细胞效力、选择性、溶解度、代谢稳定性和体内暴露量,但渗透性——乃至最终口服生物利用度——的进一步提升可能需要修饰BCL6和BRD4结合部分本身,可能需以氢键供体更少、分子量更低、极性更有利的配体替换BI-3812和JQ1。