1、摘要
本文提出含饱和杂环胺环的稠合及螺环1,4-二氧杂环庚烷可作为早期药物发现的潜在分子骨架。其合成关键步骤为合适的饱和N-杂环1,2-二醇与α,α'-二氯异丁烯的双烷基化反应;通过对所得双环中间体中端位环外双键的进一步化学修饰,获得了一系列含常见官能团(可作为合成位点)的高价值砌块。所开发的合成方案在克级规模下表现出高效性。通过理化特征与构象表征(即pKa和Log P测定、虚拟库枚举及基于X射线衍射数据的出口向量分析),评估了该类双环化合物作为等排取代基的应用潜力。
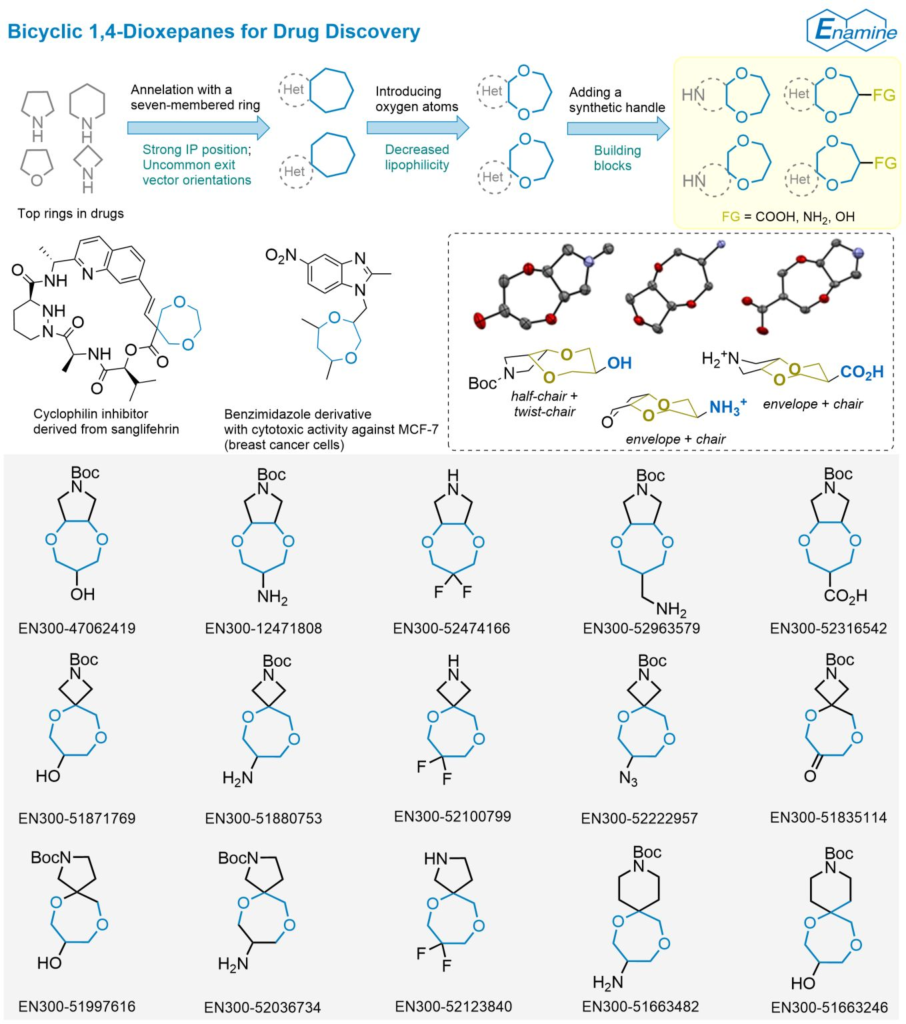
2、引言
长期以来,药物发现对新型化学型的持续需求一直是有机合成研究的主要驱动力[1]。氮杂环丁烷、吡咯烷、哌啶、四氢呋喃等饱和环在生物活性分子中广泛存在[2-4],且将这些环稠合作为药物设计通用策略已取得诸多成功案例,因此含上述片段的饱和稠合及螺环体系成为极具吸引力的合成目标[5-10]。此类双环骨架可通过增加分子三维性、改善理化性质及降低淘汰率,优化潜在药物候选物的药理特性[11,12]。
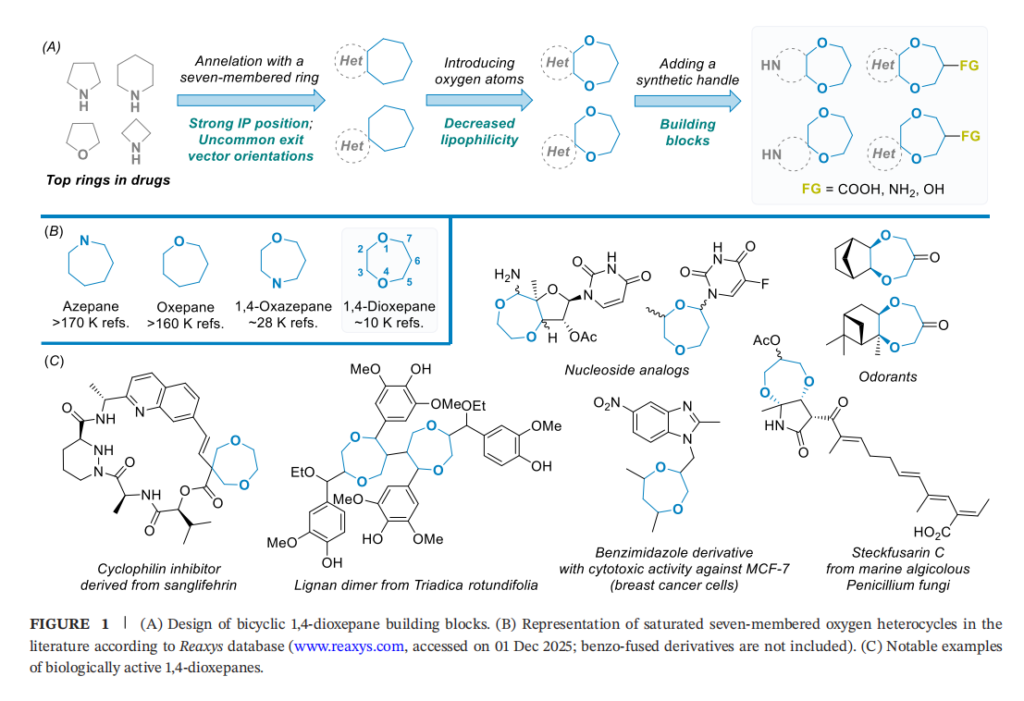
饱和七元环为新型双环体系设计提供了尚未充分探索的机会——不仅能显著提升知识产权布局优势,还能赋予连接官能团独特的相对空间取向[13,14](图1A)。该类化学型的主要问题是碳原子数增加导致脂溶性升高,而引入多个氧原子可有效缓解这一缺陷(氧原子已知能提高化合物极性)。考虑到所得环系的化学稳定性,1,4-二氧杂环庚烷衍生物尤为具有应用前景。尽管该类杂环在文献中的报道少于其他饱和七元环(图1B),但在天然产物和合成产物中均发现了众多含该结构的生物活性衍生物(图1C):例如,将1,4-二氧杂环庚烷片段引入苯并咪唑类抗肿瘤药物中,得到的化合物活性显著优于1,4-二氧六环、哌嗪及其他杂环衍生物[15];1,4-二氧杂环庚烷被用于核苷类似物设计[16],包括基于5-氟尿嘧啶的抗肿瘤药物[17];从圆叶乌桕中分离出的含两个1,4-二氧杂环庚烷环的木脂素二聚体具有中等抗神经炎症和抗氧化活性[18];通过1,4-二氧杂环庚烷螺稠合修饰桑格弗林大环,开发出生物利用度良好的亲环蛋白抑制剂[19],该修饰虽降低了膜通透性,但提升了代谢稳定性并减少孕烷X受体(PXR)激活;另一种螺环骨架——8,11-二氧杂-3-氮杂螺[5.6]十二烷被用于修饰哌啶类抗生素[20];近期从青霉菌中分离出含与吡咯烷稠合的1,4-二氧杂环庚烷环的聚酮类镰孢菌素衍生物[21];此外,双环和三环1,4-二氧杂环庚烷也应用于香料领域[22,23]。
为确保饱和双环1,4-二氧杂环庚烷在生物活性化合物发现中的适用性,需在该骨架中引入合成位点以实现高效化学修饰。对于含氮杂环,仲氨基可作为此类合成位点;同时,考虑到药物化学和组合化学中常用的官能团(NH₂、COOH、OH [24]),本研究计划将其引入1,4-二氧杂环庚烷环的C-6 位。结合分子对称性及杂原子附近部分基团(如NH₂或OH)的稳定性问题,该修饰有望简化相应衍生物的制备过程。
本研究通过开发高效的克级合成方法,制备了饱和稠合及螺环1,4-二氧杂环庚烷单功能和双功能砌块,实现了图1A 所示的设计思路。为验证合成化合物在药物化学中的应用价值(尤其是作为简单环系的潜在等排体),对其理化性质和结构特征进行了表征:测定了一系列模型衍生物的pKa和Log P值,生成虚拟化合物库并评估其类先导物特性,对部分代表性化合物进行 X 射线衍射研究,为后续构象分析和出口向量分析奠定基础。
3、结果与讨论
3.1 合成
目标双环砌块的通用合成方法应基于七元杂环的构建。由于需在1,4-二氧杂环庚烷的C-6位引入合成位点,本研究选择碱促进的1,2-二醇与α,α'-二氯异丁烯(3-氯-2-(氯甲基) 丙烯)双烷基化反应作为关键方法(方案1)[23,25-27]。该烷基化试剂不仅能提供所需的取代模式,还具有高反应活性,且不易发生消除反应(消除反应会给七元环的构建带来挑战)。此外,1,4-二氧杂环庚烷环上无官能团的目标化合物也可通过该方法制备,后续可去除非必需的双键片段。
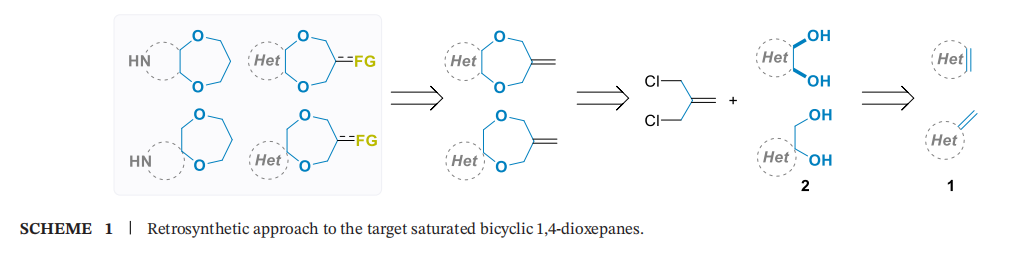
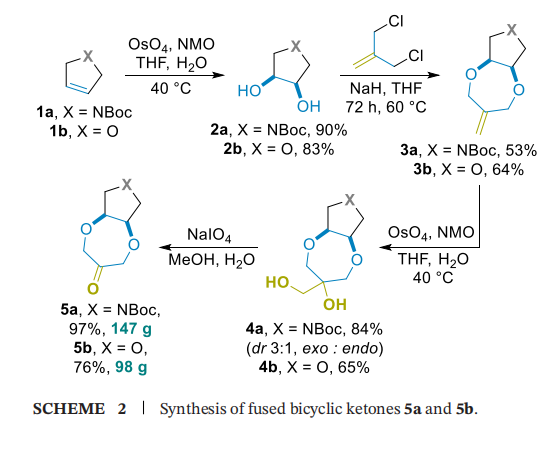
所需的杂环1,4-二氧杂环庚烷2可通过商用含环内或环外双键的烯烃1(分别用于制备稠合双环和螺环衍生物)经四氧化锇(OsO₄)介导的二羟基化反应便捷获得。
首先,采用上述合成方案制备稠合双环1,4-二氧杂环庚烷。为避免中间体手性相关的不必要复杂性,初始选择含对称五元杂环的烯烃1(即N-叔丁氧羰基吡咯啉(1a)和2,5-二氢呋喃(1b))作为底物:1a和1b经OsO₄-N-甲基吗啉-N-氧化物(NMO)顺式二羟基化反应,分别以89%和83%的产率得到已知二醇2a[28,29]和2b[30](方案2);在氢化钠(NaH)存在下,2a和2b与α,α'-二氯异丁烯发生烷基化环化反应,采用四氢呋喃(THF)替代文献报道的二甲基甲酰胺(DMF)[23,25-27] 作为溶剂,简化了分离流程;双环中间体3a和3b经OsO₄-NMO氧化环外端位双键,得到邻二醇4a和4b,再经高碘酸钠(NaIO₄)裂解,三步反应分别以43%和32%的总产率得到酮类化合物5a和5b。值得注意的是,该反应序列可实现规模化制备,5a单次反应规模可达147g,5b可达98g。需说明的是,四氢呋喃系列中间体亲水性较高,导致纯品分离难度增加,因此总产率略低,但未影响方法的整体稳健性和可重复性。
鉴于酮5a的理化性质更便于操作,以其为底物进行进一步官能化实验(方案3):经硼氢化钠(NaBH₄)还原,以60%的产率得到外消旋和内消旋非对映异构体混合物(dr=2:1)醇6a,通过甲醇重结晶分离得到主要的外消旋异构体纯品;为引入羧基(COOH),依次进行甲氧基甲基取代磷叶立德的Wittig反应、所得烯醇醚的酸性水解,以及中间体醛经高碘酸钠-三氯化钌(NaIO₄-RuCl₃)氧化,最终以34%的总产率得到羧酸8a(dr=1:2,外消旋:内消旋),该产物产率较低与Wittig反应后的纯化难度较大相关。
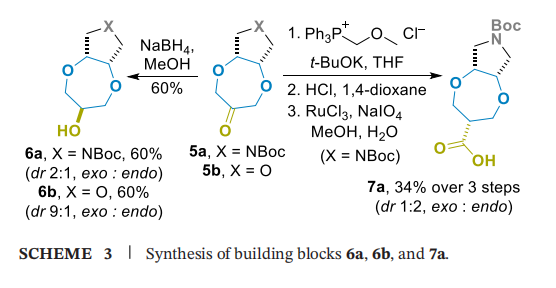
以醇6a为原料,在六氢-2H,6H-[1,4]二氧杂环庚并[2,3-c]吡咯骨架上引入氨基(NH₂)并获得母体双环体系(方案4):6a经甲磺酰化后与碘化钠(NaI)发生亲核取代反应,得到碘化物8a(分离纯化后产率30%,外消旋:内消旋=6:1);8a经催化氢解和N-叔丁氧羰基(N-Boc)脱保护,得到盐酸盐形式的胺9a(产率73%);为获得单保护二胺衍生物11a,6a经甲磺酰化转化为叠氮化物10a(产率55%,外消旋:内消旋=1:2),再经催化氢化还原,分离纯化后以85%的产率得到主要为内消旋异构体的化合物11a・HCl(dr=6:1)。
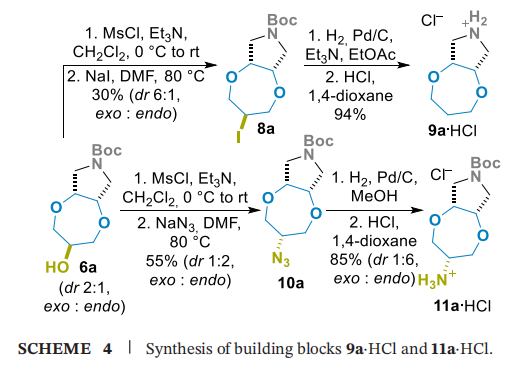
通过酮5b经NaBH₄还原,可获得规模达120g的醇6b(外消旋:内消旋=9:1,产率60%)(方案2),但该骨架亲水性高,操作难度较大,因此需开发相应羧酸7b和胺11b的替代合成路线;此外,方案3制备羧酸7a的总产率不理想,因此基于烯烃3a和3b的硼氢化-氧化反应,设计了该类化合物的替代合成方案(方案5):通过该方法得到含额外亚甲基(CH₂)的醇12a(外消旋:内消旋=9:1,产率60%)和12b(外消旋:内消旋=3:2,产率77%);12a和12b经两步氧化,分离纯化后分别以81%和90%的产率得到羧酸7a(外消旋:内消旋=7:3)和7b(外消旋:内消旋=1:1)。值得注意的是,该路线制备7a的总产率和规模化能力均优于方案3,单次反应规模可达50g。
通过羧酸7b的改良Curtius重排反应,以43%的产率得到盐酸盐形式的胺11b;此外,脱除砌块12a和7a中的N-Boc保护基,分别得到盐酸盐形式的游离氨基醇13和氨基酸14,经重结晶分离得到纯外消旋非对映异构体7b・HCl和14・HCl。
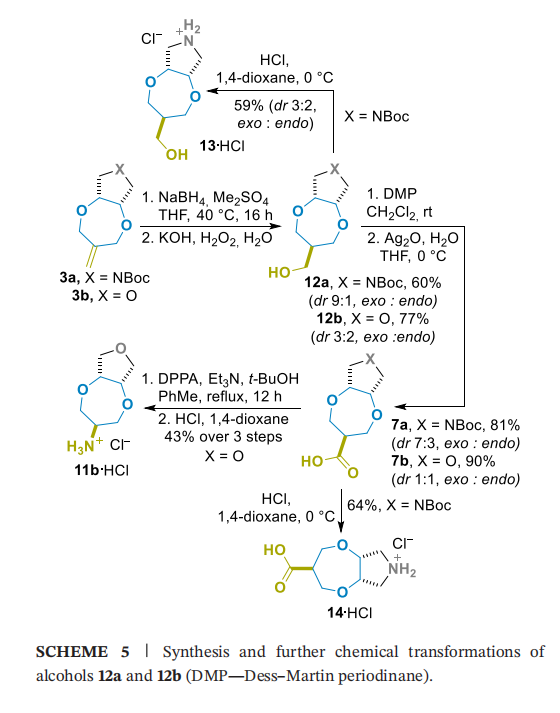
螺环1,4-二氧杂环庚烷衍生物9c-e、11c-e和15c-e由商用含环外双键的饱和杂环胺衍生物1c-e制备(方案6),反应序列与稠合型衍生物的合成策略一致:经顺式二羟基化、与3-氯-2-(氯甲基) 丙烯双烷基化(两步反应产率30%-60%,得到中间体3c-e)、两步氧化断裂双键(得到中间体5c-e,产率59%-71%);酮5c-e可作为制备母体胺9c-e(盐酸盐形式)、单保护二胺11c-e和氨基醇15c-e(盐酸盐形式)的平台,所用反应序列与方案3和4基本一致,且在克级规模下保持高效性(酮5c-e单次反应规模可达300g)。
3.2 理化性质
为评估合成的双环1,4-二氧杂环庚烷在药物发现中的潜力,测定了其简单模型酰胺衍生物的酸碱性质(pKa)和脂溶性(Log P),并与潜在等排取代目标物进行对比:含仲胺盐酸盐官能团的砌块9a-e可作为吗啉和其他饱和杂环胺的替代物;砌块7a、7b、11a和11b可用于替代天然产物中广泛存在的环烷基片段、饱和氧杂环和苯并二氧六环结构[31,32];此外,还纳入了先前报道的双环1,4-二氧六环类似物[33,34] 作为对比参照物。
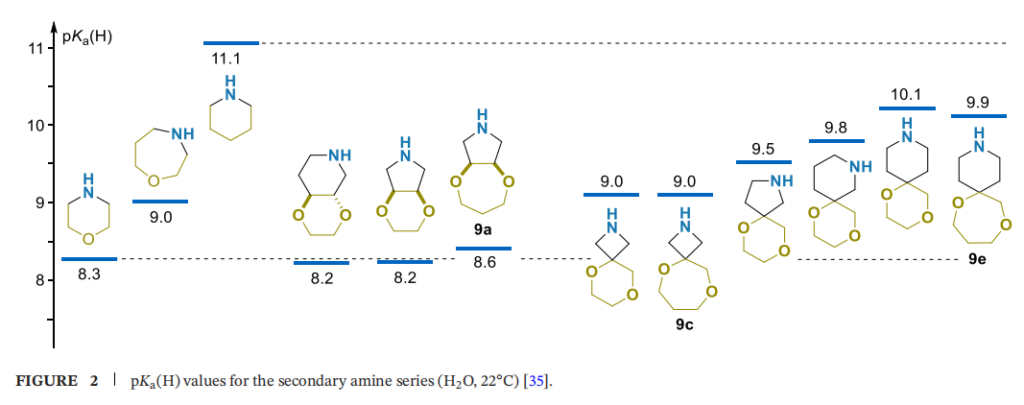
尽管上述砌块(或其简单模型衍生物)本身不太可能直接用于生物活性研究,但相关测定结果可用于评估其片段的pKa和Log P增量,进而预测相应等排取代对化合物性质的影响;此外,pKa值还可用于理解砌块的反应活性。
双环仲胺的pKa (H) 值分析表明,结构相似的1,4-二氧六环和1,4-二氧杂环庚烷衍生物碱性非常接近(图2)。该系列化合物的碱性差异与氧原子和氮原子之间的键连距离呈极佳相关性(距离越短,碱性越低),表明pKa (H) 值由杂原子的诱导效应决定:稠合双环化合物9a及其1,4-二氧六环衍生物(氮原子与两个氧原子之间均仅含三个键,即两条O-C-C-N路径)的pKa值介于8.2-8.6之间,与吗啉(pKa (H)=8.3)接近,因此9a及其类似物可作为吗啉的潜在替代物。
螺环化合物9c-e及其相应1,4-二氧六环类似物的pKa值介于9.0-10.1之间,处于吗啉(pKa (H)=8.3)和哌啶(pKa (H)=11.0)之间,且与广泛使用的吗啉同系物1,4 - 氧氮杂环庚烷(pKa (H)=9.0)非常接近。
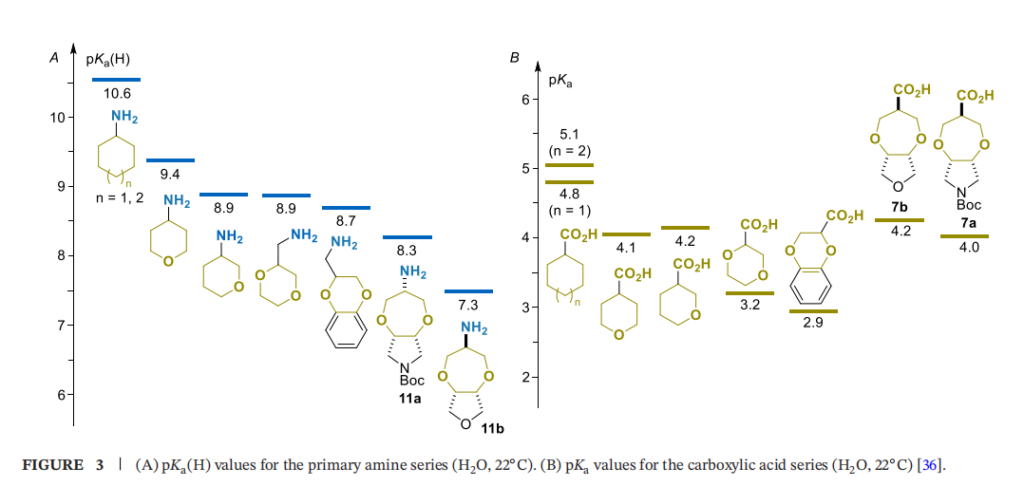
伯胺11a和11b的碱性较环己烷/环庚烷(pKa (H)=10.6)、四氢吡喃(4-异构体和3-异构体的pKa (H) 分别为9.4和8.9)以及同源1,4-二氧六环和1,4-苯并二氧六环(pKa (H) 分别为8.9和8.7)显著降低(图3A),该现象可通过氧原子的诱导效应合理解释:11a和11b分子中存在两条最短的N-C-C-O路径,而1,4-二氧六环和四氢吡喃-3-基衍生物中仅存在一条,其余氧原子与氮原子的键连距离均较长。11a和11b之间pKa值相差1.0个单位的原因尚不明确,可能与其构象特征相关(见下文)。
羧酸7a和7b的酸性趋势同样与氧原子的诱导效应(及氧原子与羧基(CO₂H)的键连距离)相关,但该系列化合物的pKa差异较不显著(图3B):具有相同立体化学的7a和7b酸性相似(pKa分别为4.0和4.2),与四氢吡喃衍生物(pKa=4.1-4.2)接近。
Log P测定中,图2所示胺类经标准苯甲酰化反应转化为相应苯甲酰胺16a-h(详见补充信息)。选择苯甲酰胺衍生化是基于测定方法的特定需求:便于串联质谱定量分析和结果解读,同时可将Log P值调整至更合适的范围。结果表明,尽管合成的模型双环衍生物含更多非氢(重)原子,但除个别情况外,其脂溶性均低于N-苯甲酰哌啶,且高于N-苯甲酰吗啉,因此该类化合物可作为哌啶或吗啉的潜在等排体,用于精细调控化合物的脂溶性。
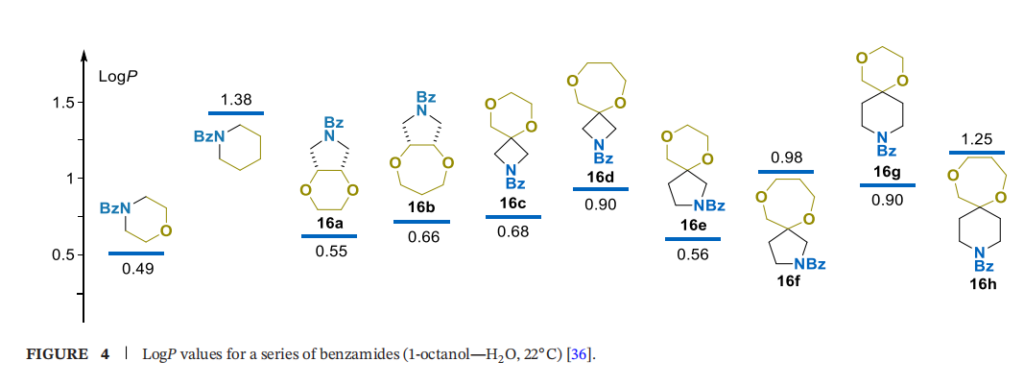
在双环衍生物16a-h系列中,1,4-二氧杂环庚烷衍生物的脂溶性比相同取代模式的1,4-二氧六环衍生物高0.1-0.4个单位,这与额外亚甲基的作用相关;该差异受酰胺片段与氧原子之间距离的影响:距离越短,亚甲基的脂溶性效应越弱,可能是由于吸电子基团对亚甲基的极化作用更强。
几乎所有情况下,Log P值与主骨架的重原子数(HAC)呈良好正相关,每个亚甲基平均使Log P值升高0.2个单位。化合物16e的脂溶性低于16c和16g的原因尚不明确,可能与分子对称性较低导致偶极矩较高相关;重原子数相同的情况下,稠合衍生物16a和16b的脂溶性分别低于其螺环类似物16c和16d。
3.3 类药性与类先导物特性评估
为进一步评估合成的砌块在生成具有药物化学相关理化性质化合物方面的潜力,生成了其衍生物小型库并评估类先导物特性。采用Nelson课题组报道的方法[37],使用一组标准化反应和修饰反应物(结构详见补充信息),对用于Log P实验测定的胺类化合物进行虚拟衍生;通过Instant JChem软件的标准工具进行虚拟偶联和理化性质预测[38],最终得到含240个成员的库,其中包括96个1,4-二氧杂环庚烷衍生物(图5)。
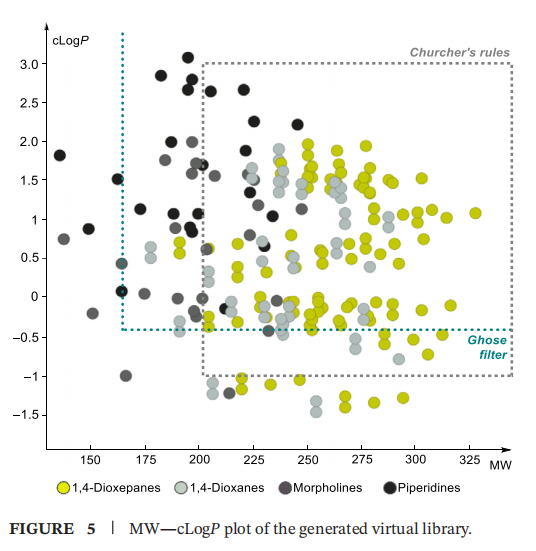
所有生成的库成员均符合常见类药性准则,包括Lipinski五规则(分子量MW<500、Log P≤5、氢键供体数HDon≤5、氢键受体数HAcc≤10)[39]和穆格药效团筛选器(Muegge pharmacophore-based filter)[40];戈斯筛选器(Ghose filter)(MW=160-480、Log P=-0.4-5.6、原子数AC=20-70、分子折射率MR=40-130)筛除了42个成员(占18%),其中14个为1,4-二氧杂环庚烷衍生物,证实了生成库成员的类药性。此外,全库均符合奥普雷亚课题组提出的类先导物四规则(Oprea's "rule of four")(MW<400、Log P≤4、HDon≤4、HAcc≤8)[41];即使是最严格的丘彻类先导物准则(Churcher's lead-likeness rules)(MW=200-350、Log P=-1-3、无反应性基团)[42],240个库成员中也有179 个(占75%)符合,其中1,4-二氧杂环庚烷衍生物子集的达标率为86%(83/96)。此外,1,4-二氧杂环庚烷衍生物子集的平均预测脂溶性(cLog P=0.5)低于哌啶(cLog P=1.6),且与吗啉(cLog P=0.6)和1,4 - 二氧六环(cLog P=0.4)衍生物非常接近。上述结果表明,与模型苯甲酰胺的实验数据相比,Log P预测算法倾向于低估双环1,4-二氧杂环庚烷的脂溶性。
3.4 结构分析
为评估目标化合物的构象行为,对三个代表性化合物(6a、11b・HCl・H₂O和14・HCl)进行X射线衍射研究(图6):化合物11b・HCl・H₂O中,1,4-二氧杂环庚烷环采取椅式构象,O-C-C-O二面角为4°[13];四氢呋喃环采取信封式构象,C-C-C-C二面角为3°[43];五元环的氧原子呈内式折叠,质子化氨基取代基处于假平伏位。
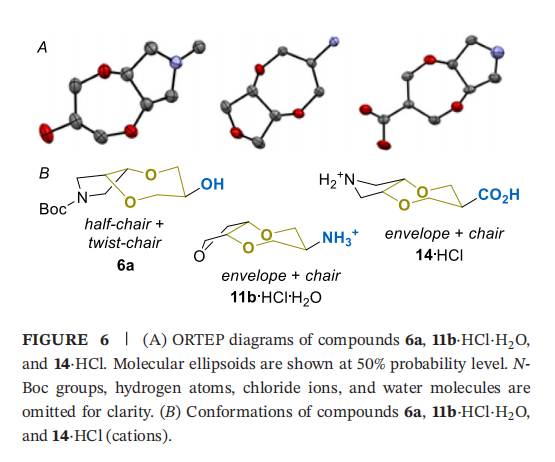
氨基酸盐酸盐14・HCl中,七元环和五元环分别采取椅式和信封式构象,相应二面角分别为11°(O-C-C-O)和9°(C-C-C-C),表明上述典型构象存在一定扭曲;但吡咯烷环的质子化氮原子呈外式折叠,导致分子整体几何结构不同;与前述情况一致,羧基处于假平伏位。
化合物6a表现出不同的构象行为:七元环和五元环分别采取扭椅式和半椅式构象,O-C-C-O 和 C-C-C-C 二面角分别为38°和36°,表明分子具有更高的整体三维性;羟基同样处于假平伏位。
化合物11b・HCl和6a环系的构象差异可能是11b和11a酸碱性质不同的原因,但需谨慎解读该结果——数据表明所讨论的双环体系具有一定构象柔性,因此6a和11a的环系构象可能不同。
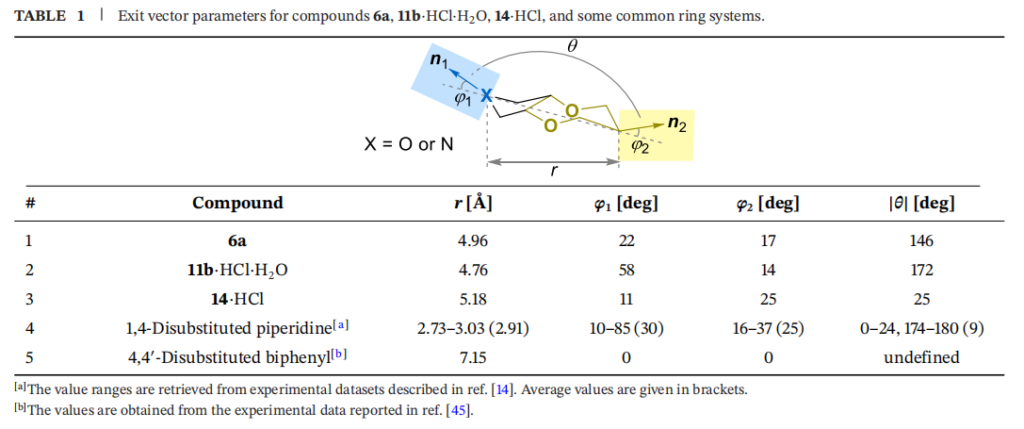
描述两个出口向量n₁和n₂的相对取向需四个几何参数:起点间距离r、平面角φ₁和φ₂以及二面角θ。表1列出了从X射线衍射数据中获取的化合物6a、11b・HCl・H₂O和14・HCl的这些参数定义及数值。为确定双环1,4-二氧杂环庚烷衍生物提供的官能团相对取向,并与其他环系进行对比,采用出口向量分析:该方法中,双功能骨架上的两个官能团通过出口向量 n₁和 n₂模拟[14,44](表1);若官能团为环系的一部分(如环状胺或醚),可通过C-X-C价键角的角平分线定义相应出口向量[14]。
结果表明,6a和14a・HCl分子中含有的3,7-二取代顺式-六氢-2H,6H-[1,4]二氧杂环庚并[2,3-c]吡咯骨架,其角参数与1,4-二取代哌啶相似(二面角θ的偏差最大),但尺寸约为哌啶的1.7倍(r=4.76-5.18 Å,哌啶r=2.91 Å)。因此,所讨论的双环1,4-二氧杂环庚烷骨架可作为加长型且轻微扭曲的哌啶替代物;顺式-六氢-2H,6H-[1,4] 二氧杂环庚并[2,3-c]吡咯骨架的r值范围介于1,4-二取代单环体系和4,4'-二取代联苯(7.15 Å)[45] 之间。
4、结论
本研究开发了基于饱和N/O杂环邻二醇与α,α'-二氯异丁烯双烷基化反应的稠合双环和螺环1,4-二氧杂环庚烷通用合成方案。所得双环体系中的环外双键可进行多样的化学选择性和区域选择性官能化,获得一系列对合成化学和药物化学有价值的单功能和双功能砌块(羧酸、胺、醇、酮等)。该方法具有高效规模化特性,稠合型衍生物单次反应规模可达150g,螺环型可达300g。
含环内氮原子的合成系列衍生物(即二氧杂氮杂螺[n.m]烷和二氧杂氮杂双环[n.m.0]烷)可作为饱和杂环胺(如哌啶和吗啉)的潜在等排取代基。模型衍生物的pKa和Log P测定表明,该类双环体系的碱性和脂溶性介于哌啶和吗啉之间,可实现生物活性分子中这些性质的精细调控。其他合成的双环1,4-二氧杂环庚烷衍生物可用于替代环烷基片段、饱和氧杂环和苯并二氧六环结构,理化表征结果也证实了这一点。基于合成的氮杂双环骨架生成的虚拟库表明其符合类先导物准则。最后,X射线衍射研究表明,二氧杂氮杂双环[n.m.0]烷骨架可作为加长型且轻微扭曲的哌啶替代物;固态下双环1,4-二氧杂环庚烷体系表现出一定构象柔性。
本文中涉及的含1,4-二氧杂环庚烷砌块与相关衍生物,Enamine均有现货供应,如需了解更多可访问官网查询:https://www.enamine-genez.com/