摘要
硫羰基叶立德CH2=S(+)−CH2(−)与缺电子环烯烃的[3+2]环加成反应可制得双环硫醚类化合物,该反应在毫克、克乃至数克级反应规模下均能高效进行。对所得产物进行标准修饰,可得到适用于药物化学研究的稠合与螺环砜类化合物。
引言
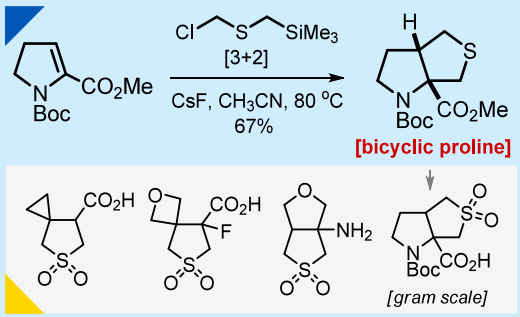
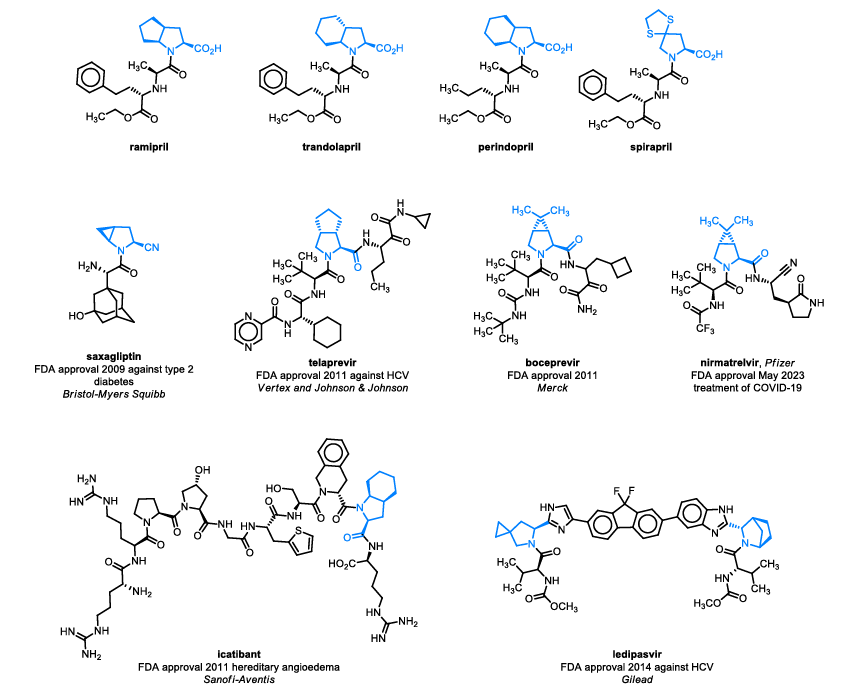
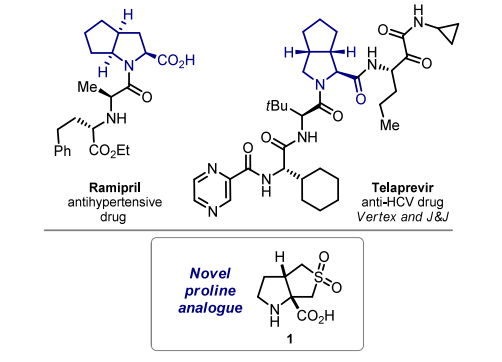
双环脯氨酸是现代药物研发中应用广泛的合成砌块,雷米普利、特拉普韦等药物的结构中均含有该类基团。基于对这类分子的持续研究兴趣,本研究计划合成新型含SO2的双环脯氨酸1(图1)。本文将报道该化合物的合成方法,并阐述关键的[3+2]环加成步骤如何逐步发展为合成药物化学用稠合与螺环砜类化合物的通用方法。
对氨基酸1进行逆合成分析后,我们推测烯烃2可能也能与硫羰基叶立德发生类似反应。1986年,Sakurai课题组发现,硫化物3在氟化铯存在下可生成硫羰基叶立德CH2=S(+)−CH2(−),且该叶立德能与直链缺电子烯烃发生反应。本研究尝试将该反应应用于烯烃2,最终成功实现目标产物的合成。
反应条件优化
此前研究发现,烯烃2可与N-苄基甲亚胺叶立德发生[3+2]环加成反应。本研究以该反应为基础,对烯烃2与硫化物3的[3+2]环加成反应条件进行优化,结果如表1所示。
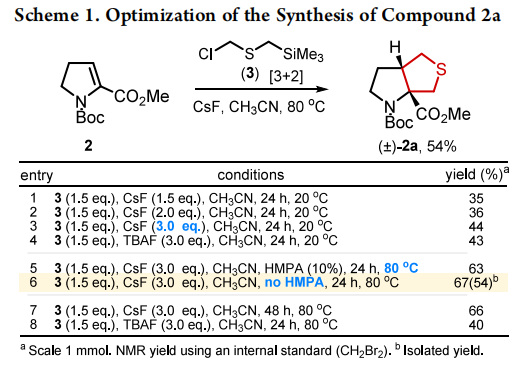
实验结果表明,20℃下增加氟化铯用量至3.0当量,产物2a的收率可从35%提升至44%;将反应温度升至80℃,即使不加六甲基磷酰胺,收率也能达到67%(分离收率54%),说明六甲基磷酰胺对该反应收率无显著影响;使用四丁基氟化铵替代氟化铯,收率未得到提升。最终确定最优反应条件为:硫化物3(1.5当量)、氟化铯(3.0当量)、乙腈为溶剂,80℃反应24h。
氨基酸1的合成
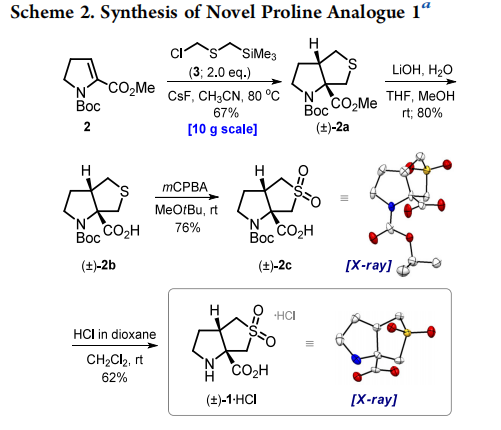
在上述优化后的反应条件下,烯烃2向2a的转化反应可放大至10克级,仅需将硫化物3的用量增至2.0当量。经柱层析纯化后,产物2a的分离收率为67%。
对2a进行酯基皂化得到2b,再对硫原子进行氧化得到N-保护的氨基酸2c;最后通过标准的酸性脱N-叔丁氧羰基保护基反应,得到新型脯氨酸类似物1的盐酸盐1・HCl。X射线单晶衍射分析确证了化合物2c和1・HCl的结构(图中碳原子为灰色、氧原子为红色、氮原子为蓝色、硫原子为黄色,椭球概率水平为40%,为清晰起见,氢原子和氯原子已省略)。
反应适用范围
砜基是有机合成和药物化学中常见的官能团,基于此,本研究进一步探索了该[3+2]环加成策略在合成其他官能化双环砜类化合物中的应用,分别考察了稠环产物和螺环产物的合成效果,并分析了反应的局限性及解决方法。
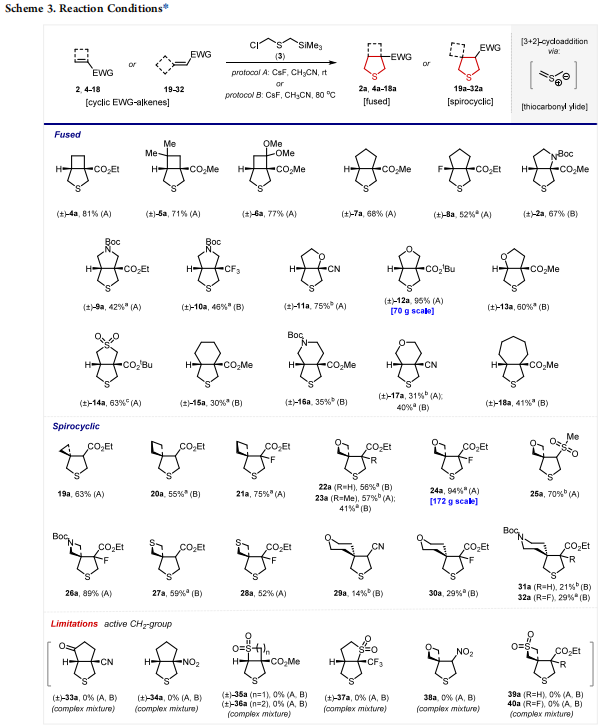
稠环产物
环内四元烯烃4~6在室温下可与硫化物3顺利反应,以71%~81%的收率得到产物4a~6a;五元烯烃7和8的反应收率为52%~68%;杂原子取代的五元烯烃9~14也能与3发生反应生成9a~14a,其中钝化烯烃10(含大位阻三氟甲基)和13(推-拉电子型烯烃)的反应速率较慢,需加热并补加试剂才能完成反应;六元及七元烯烃15~18的反应活性最低,产物15a~18a的收率中等,仅为30%~41%。
螺环产物
环外三元及四元烯烃19~28的反应性整体较好,产物19a~28a的收率为52%~94%。值得注意的是,在烯烃片段中引入氟原子(21、24、26)可显著提高其反应活性,促进反应进行;六元烯烃29~32的反应活性较差,螺环四氢噻吩类化合物29a~32a的收率仅为14%~29%。
反应局限性
含活性亚甲基的烯烃33~40参与反应时,会生成复杂的混合物。推测其原因是氟化铯作为碱,会使含活性亚甲基的底物发生脱质子反应并进一步分解,导致反应无法定向进行。
局限性解决方法
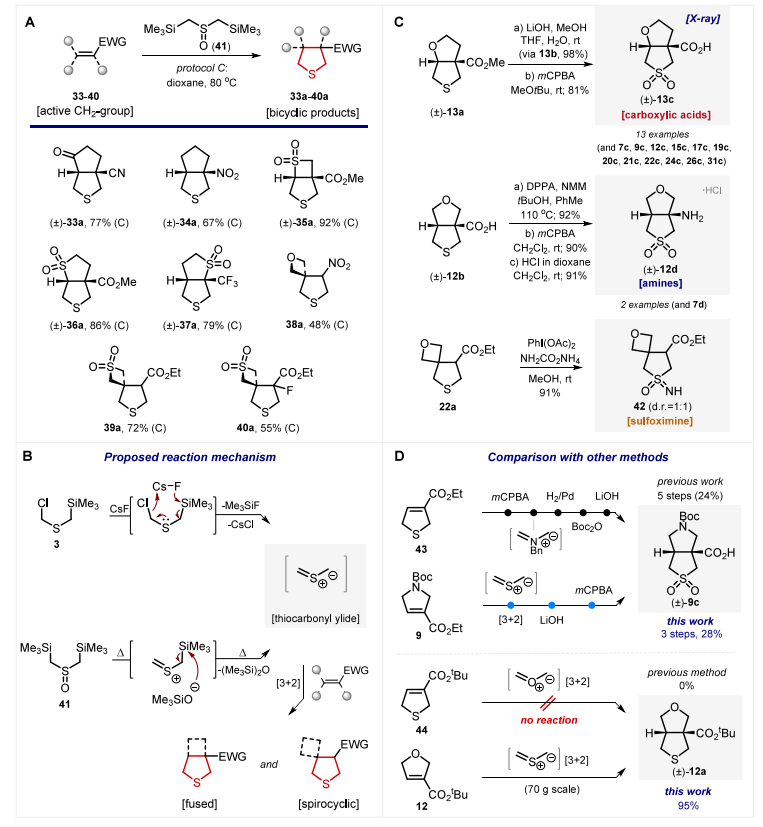
1986 年,Achiwa课题组报道,亚砜41在六甲基磷酰胺中加热,无需氟化铯即可生成硫羰基叶立德CH2=S(+)−CH2(−)。本研究借鉴该方法,将烯烃33~40与亚砜41在1,4 - 二氧六环中加热反应(无六甲基磷酰胺),成功实现了目标产物的合成,此前难以制备的产物33a~40a均能快速生成,收率为48%~92%。
反应机理
本研究提出了该[3+2]环加成反应的反应机理(方案4B):
| 1 | 以硫化物3为原料时,氟离子进攻硅原子,生成硫羰基叶立德CH2=S(+)−CH2(−); |
| 2 | 以亚砜41为原料时,加热引发三甲基硅氧基阴离子的热消除,该阴离子进一步进攻另一个硅原子,最终生成硫羰基叶立德CH2=S(+)−CH2(−); |
| 3 | 硫羰基叶立德与缺电子环烯烃发生非对映选择性[3+2]环加成反应,生成最终的稠合与螺环产物。 |
产物修饰
对上述环加成产物(通式a)进行酯基/氰基水解得到羧酸类化合物(通式b),再对硫醚片段进行氧化,可得到含砜基的羧酸类化合物(通式c);对羧酸b进行Curtius重排反应,随后氧化硫醚并脱N - 保护基,可制得双环胺类化合物7d和12d;硫化物22a与二乙酸碘苯在氨基甲酸铵存在下反应,能以91%的收率生成硫代亚胺砜42。
与其它方法的对比
此前有研究以烯烃43为原料,经五步反应以24%的收率得到氨基酸9c;而本研究以烯烃9为原料,仅经三步反应就以28%的收率合成了9c,步骤更短、收率更高。
对化合物12a进行基于[3+2]环加成的逆合成分析,有两种可行策略:第一种是烯烃44与非稳定羰基叶立德反应,但本研究在已报道的反应条件下尝试该转化时,未观察到反应发生,反应体系中仅存在起始烯烃;第二种是烯烃12与硫羰基叶立德反应,该反应能顺利进行,且可放大至70克级,目标产物12a的收率高达95%,体现了本研究方法的优越性。
结论
硫羰基叶立德CH2=S(+)−CH2(−)与缺电子环烯烃的[3+2]环加成反应可高效制备稠合与螺环硫醚类化合物。根据起始烯烃的结构差异,可灵活选用硫化物3或亚砜41作为硫羰基叶立德前体进行反应。对所得硫醚产物进行标准的有机合成修饰,能够得到一系列适用于药物化学研究的稠合与螺环砜类化合物,为药物研发提供了新型结构多样的合成砌块。
本文中涉及的含稠合与螺环砜类化合物砌块与相关衍生物,Enamine均有现货供应。