编者按:Enamine虚拟库以REAL Space为核心,坐拥超820亿种可快速合成的枚举化合物,依托超31万种现货分子砌块、180+验证合成方案实现3-4周平行合成且成功率超80%,还可结合MADE库实现4-8周定制合成,化学空间庞大、合成可行性高、交付速度快,能全方位支撑苗头化合物扩展与先导化合物优化。此文探讨了REAL Space文库对接技术在双靶点联合活性筛选中的应用
1、摘要
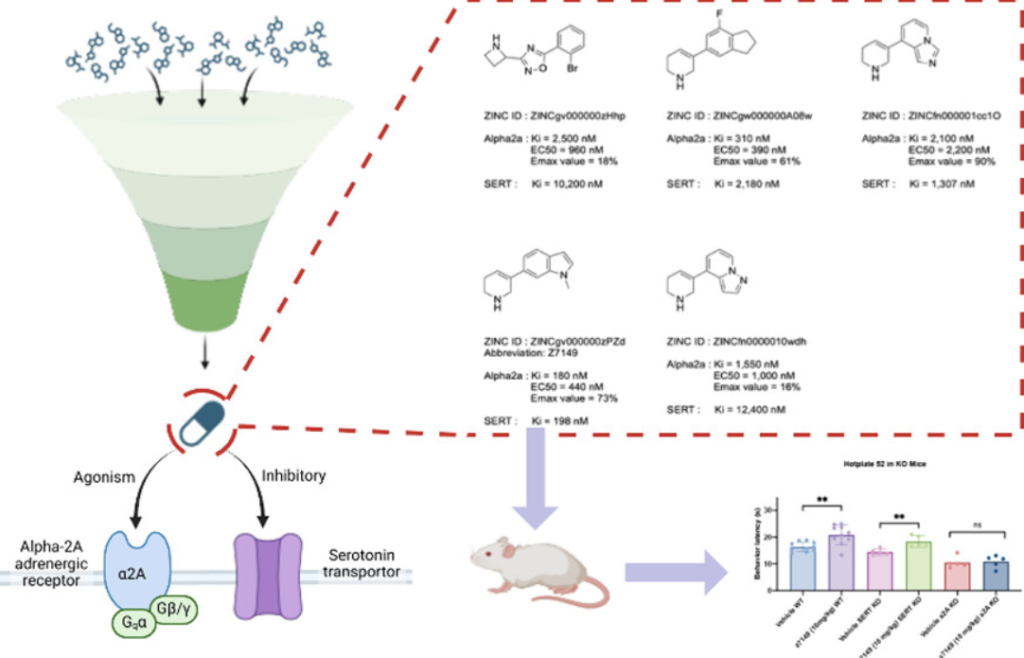
多靶点药物分子对治疗复杂疾病具有重要吸引力。本研究探索了大型文库对接技术在双靶点联合活性筛选中的应用。回顾性研究表明,随着文库规模扩大,潜在的双活性分子数量也随之增加。在针对三组靶点对(α₂ₐ肾上腺素能受体/血清素转运体、μ-阿片受体/血清素转运体、α₂ₐ肾上腺素能受体/μ-阿片受体)的前瞻性对接中,我们对含9亿个分子的文库进行筛选,旨在发现镇痛化合物。α₂ₐ/血清素转运体和μ-阿片受体/血清素转运体两组筛选均获得了具有低微摩尔至高中纳摩尔活性的双靶点结合物,且命中率较高;其中α₂ₐ/血清素转运体筛选得到的四氢吡啶类化合物还对5-羟色胺₂ₐ受体具有活性。尽管冷冻电镜结构证实了对接预测的结合构象,但在优化过程中难以提高化合物的效价。不过,在小鼠行为学实验中,活性最强的α₂ₐ/血清素转运体双靶点化合物(z7149)展现出有效的镇痛作用,且未诱导条件性位置偏爱,同时具有强效的抗抑郁和抗焦虑类药物活性,这与其血清素转运体/5-羟色胺₂ₐ受体的双重活性一致。本研究揭示了对接技术在多靶点药理学研究中的优势与挑战。
2、引言
多药联用存在诸多局限性,这促使研究人员探索多靶点药理学——即单个分子同时调控多个靶点的研究方向⁴。多靶点作用在有效药物中十分常见,例如抗精神病药氯氮平,它既作用于D₂多巴胺受体以治疗精神分裂症的阳性症状,又作用于5-羟色胺₂ₐ受体以缓解纯D₂家族拮抗剂(如氯丙嗪或氟哌啶醇)引起的运动障碍⁵。类似地,三重再摄取抑制剂(如LPM580098⁶,⁷)因相较于选择性再摄取抑制剂具有更高疗效,已被开发用于治疗神经性疼痛。在分子层面,通路分析阐明了作用于激酶级联反应多个位点的药物具有更高疗效和更低毒性的机制⁸,⁹。此外,替尔泊肽对葡萄糖依赖性促胰岛素多肽受体和胰高血糖素样肽-1受体的双重活性¹⁰⁻¹²,可能提高其治疗肥胖症和2型糖尿病的疗效;该领域是意向性G蛋白偶联受体多靶点药理学研究中最活跃的方向之一。然而,除少数概念验证研究外⁸,¹³,大多数多靶点药物并非从头设计,其多靶点作用往往在药物获批后才被发现¹⁴⁻²⁰。
许多疾病由多种蛋白质共同驱动,联合靶向这些蛋白质可能提高治疗效果。例如,血压的充分控制通常需要联合使用作用于钙通道的药物、作用于G蛋白偶联受体(如血管紧张素受体、β₂和α₂肾上腺素能受体)的药物,以及作用于血管紧张素转换酶的药物。在癌症治疗中,激酶抑制剂常与靶向DNA复制和修复的药物联合使用;而作用于环氧合酶的抗炎药则常与μ-阿片受体激动剂联合用于镇痛。针对细菌感染,靶向核苷生物合成途径不同步骤的药物(如磺胺类药物和甲氧苄啶),以及氨基糖苷类与β-内酰胺类等组合,可产生协同作用。这些药物通常以固定剂量组合形式使用,以提高治疗效果、安全性或患者依从性¹⁻³。但多药联用存在个体药物药代动力学差异、制剂工艺复杂以及药物相互作用等问题¹,²。
目前已有多种方法用于意向性设计或发现多靶点配体,包括基于配体的方法、基于行为的筛选⁴,²¹⁻³³ 以及基于结构的设计¹³,²⁴。本研究聚焦于文库对接技术,该技术的应用效果参差不齐。尽管已有通过对接筛选意向性调控多个靶点的分子的案例²³,²⁵,²⁸,²⁹,但这些分子对多个靶点的亲和力通常较低,或仅能作用于亲缘关系较近的靶点。同时,寻找兼具正确功能(如激动剂或拮抗剂)的双靶点配体也具有不确定性。尽管我们此前已成功发现针对两个受体的双靶点分子,但这些靶点通常亲缘关系较近(如多巴胺D₂与血清素5-羟色胺₂ₐ受体、μ-与κ-阿片受体)³⁴,³⁵,且难以获得具有预期功能的配体³⁴,³⁵,其多靶点作用往往与非特异性相关³⁴。在一项寻找D₂和5-羟色胺₂ₐ受体联合拮抗剂的研究,以及一项寻找μ和κ-阿片受体激动剂/拮抗剂的研究中³⁵,我们发现百万级规模的对接文库可能不足以筛选出支持选择性多靶点作用的配体。
本研究首先探究了文库规模扩大对基于对接的双靶点配体发现的影响。在回顾性研究中,我们分析了文库规模增长对筛选已知及类已知多靶点配体能力的作用。这些研究为前瞻性对接筛选提供了支持,旨在发现针对不同蛋白质家族靶点对的配体,包括G蛋白偶联受体家族的α₂ₐ肾上腺素能受体与血清素转运体,以及血清素转运体与G蛋白偶联受体家族的μ-阿片受体(二者联合用于镇痛)。随后,我们通过实验验证了这些筛选预测的分子对成对靶点的联合活性。此外,我们还分析了这些研究的总体命中率、多靶点配体的效价和药代动力学优化能力,以及配体实验结构与预测构象的一致性,同时评估了其作为非强化性镇痛剂、抗抑郁药和抗焦虑药的体内活性。本研究还探讨了该方法的机遇与重大挑战。
3、结果
已知多靶点靶点对的回顾性配体富集
我们选取了三组已知共享配体且具有可用结构的受体对,这些受体对均已在单个靶点对接中获得成功³⁴,³⁶⁻³⁹,包括:D₂多巴胺受体与5-羟色胺₂ₐ血清素受体、μ-阿片受体与κ-阿片受体、血清素转运体与多巴胺转运体。
我们首先以与已知双靶点配体的相似性为指标,探究了三组靶点对的潜在双靶点配体数量随文库规模的变化规律。以D₂和5-羟色胺₂ₐ受体对为例,我们从ChEMBL数据库中筛选出177种对两个靶点的亲和力均优于1μM的已知配体,并利用ECFP4拓扑指纹和塔尼莫托系数(Tc)计算了这些配体与ZINC22数据库中30亿个化合物的相似性⁴⁰。为评估文库规模的影响,我们从30亿个化合物中随机选取规模为1000、10,000…… 直至10亿个化合物的子集,重复100次并绘制相似性分布。结果显示,随着文库规模扩大,发现与已知双靶点化合物相似分子的概率也随之增加(图1A;补充信息图1)。
在证实潜在双靶点配体数量随文库规模增长后,我们进一步探究了对接技术是否能相对于文库中其他化合物(视为非结合物)富集这些配体及其类似物。我们从ZINC22数据库中随机选取不同规模的文库,采用此前针对这些靶点的对接参数³⁸,⁴¹,对D₂和5-羟色胺₂ₐ受体进行对接。筛选对接得分排名前0.05%的分子,并识别同时出现在两个靶点筛选列表中的化合物。结果表明,随着文库规模扩大,对接技术从两个受体的前0.05%排名分子中发现类已知多靶点配体的概率也增加(图1B)。当文库规模达到10亿个化合物时,对接技术从高排名分子中发现了数百种潜在的多靶点分子(补充信息图2-4)。
新型多靶点激动剂的前瞻性发现
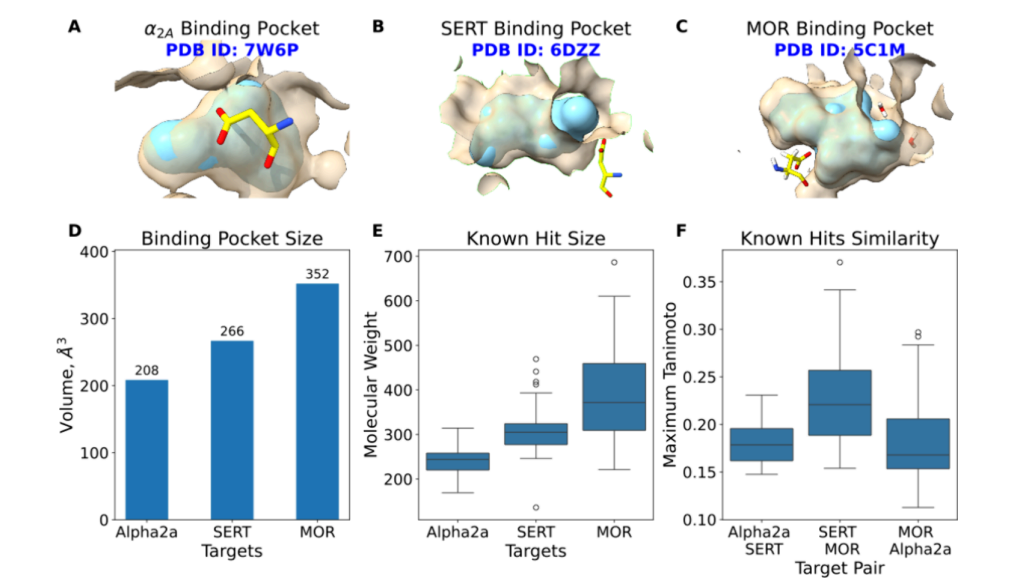
在回顾性研究结果的鼓舞下,我们选取了三个与疼痛或抗抑郁样作用相关、但结构不同的靶点:α₂ₐ肾上腺素能受体、血清素转运体和μ-阿片受体。α₂ₐ和μ-阿片受体激动剂均具有明确的镇痛作用³⁷,⁴²⁻⁴⁸,且联合使用时效果具有协同性⁴⁹⁻⁵¹。同时,血清素转运体抑制剂被认为可用于治疗慢性和神经性疼痛⁵²⁻⁵⁷,但其单独使用时疗效通常有限。本研究旨在寻找具有以下功能的分子:(1)激活α₂ₐ肾上腺素能受体并抑制血清素转运体;(2)激活μ-阿片受体并抑制血清素转运体;(3)联合激活α₂ₐ肾上腺素能受体和μ-阿片受体。我们的策略与回顾性研究一致,即寻找在成对靶点中均排名靠前的对接分子,这一目标具有挑战性但具备可行性。其中α₂ₐ/血清素转运体和μ-阿片受体/血清素转运体两组靶点对涉及不同折叠家族的靶点,而μ-阿片受体/α₂ₐ肾上腺素能受体组的正构位点大小存在差异,但三组靶点对均具有配体识别共性,包括与阳离子配体基团相互作用的关键天冬氨酸,以及识别配体芳基的芳香族残基区域(图2)。
对接参数与此前的单靶点研究保持一致³⁶,³⁷,⁴⁸,对接分子来自ZINC22数据库的定制合成分子。沿用此前研究的采样和评分方法具有双重优势:一方面可利用已成功的对接筛选经验(这些筛选通常具有较高命中率和较强活性的对接产物),另一方面便于比较单靶点对接与本研究双靶点对接的结果。
简要来说,针对μ-阿片受体/血清素转运体靶点对,我们对约9亿个阳离子分子进行了超过3万亿种构象的对接;而由于α₂ₐ肾上腺素能受体口袋尺寸限制,针对α₂ₐ/血清素转运体和α₂ₐ/μ-阿片受体靶点对,我们仅对3300万个类片段阳离子分子进行了约500亿种构象的对接。所有对接均使用DOCK 3.8软件,并采用其基于物理的评分函数。从每个筛选中收集排名前100万的化合物,分别为α₂ₐ/血清素转运体、μ-阿片受体/α₂ₐ肾上腺素能受体、μ-阿片受体/血清素转运体靶点对识别出110,000、117,000和500,000个共享化合物。通过排除构象应变较大、对接复合物中氢键供体未饱和或缺失关键相互作用(如与保守天冬氨酸形成3.2Å以内的离子对)的分子后,三组靶点对分别剩余908、819和280种潜在的多靶点化合物。通过视觉inspection最终筛选出用于合成和测试的化合物(图3)。
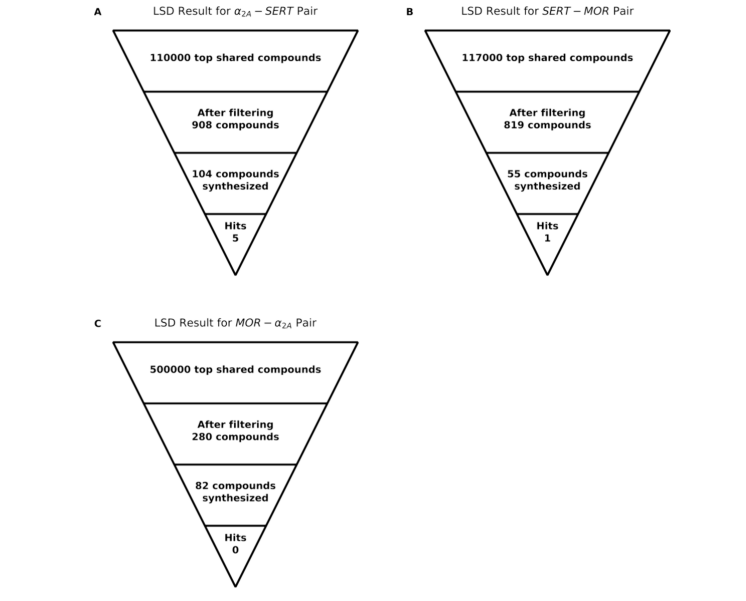
最终,针对α₂ₐ/血清素转运体、μ-阿片受体/血清素转运体、μ-阿片受体/α₂ₐ肾上腺素能受体靶点对,分别从9亿个对接化合物中选取了104、55和82种排名靠前的化合物进行从头合成和测试(表1)。所选化合物在各自的对接筛选中均排名前0.1%。由于对接化合物数量庞大,化合物在两个靶点中的排名存在差异:α₂ₐ/血清素转运体组中,α₂ₐ肾上腺素能受体的排名范围为16-996,834,血清素转运体的排名范围为2632-995,729;μ-阿片受体/α₂ₐ肾上腺素能受体组中,α₂ₐ肾上腺素能受体的排名范围为2912-113,673,μ-阿片受体的排名范围为2569-994,834;μ-阿片受体/血清素转运体组中,血清素转运体的排名范围为2-997,841,μ-阿片受体的排名范围为392-998,570。在寻找双靶点配体的约束条件下,我们接受的配体百分位排名低于单靶点筛选³⁶,³⁷,⁴⁸。
首先在10μM浓度下对α₂ₐ肾上腺素能受体、30μM浓度下对血清素转运体、100μM浓度下对μ-阿片受体进行放射性配体置换实验。对于在这些浓度下置换率超过50%的化合物,进一步测定完整的浓度-反应曲线(补充信息图5)。对于表观抑制常数(Ki)<5μM的化合物,进行α₂ₐ肾上腺素能受体和μ-阿片受体的功能研究(补充信息图6和7)。采用生物发光共振能量转移Gᵢ激活实验评估α₂ₐ肾上腺素能受体的激动活性,采用GloSensor环磷酸腺苷积累实验评估μ-阿片受体的激动活性,均进行浓度-反应分析(方法部分)。
我们此前已对α₂ₐ肾上腺素能受体、血清素转运体和μ-阿片受体进行过单靶点对接³⁶,³⁷,⁴⁸,因此比较了原始单靶点对接与本研究双靶点对接的实验验证命中率(活性化合物数量/实验测试化合物数量)。由于增加了需在两个靶点中均排名靠前的约束条件,预期命中率会下降,但实际情况并非总是如此。在α₂ₐ/血清素转运体筛选中,血清素转运体的命中率从单靶点对接的36%³⁶降至20%;但在μ-阿片受体/血清素转运体的多靶点筛选中,血清素转运体的命中率升至49%。在μ-阿片受体/α₂ₐ肾上腺素能受体筛选中,两个靶点的命中率均低于单靶点对接,但与血清素转运体联合筛选时,命中率与单靶点对接相近(表1)。尽管命中率比较需谨慎(筛选流程中化合物最终选择存在人为因素),但高于预期的双靶点命中率可能与本研究使用的文库规模大于原始研究有关,因为命中率通常随文库规模扩大而提高⁵⁹,⁶⁰。事实上,文库规模对命中率的影响正是本研究的动机之一。
尽管单个靶点的命中率通常得以维持,但双活性配体的效价通常低于单靶点对接筛选的化合物。例如,单独靶向α₂ₐ肾上腺素能受体时,最强效配体为10nM的激动剂⁴⁸,而本研究中最强效的α₂ₐ肾上腺素能受体激动剂为390nM;单独靶向血清素转运体时,最强效抑制剂的Ki值为29nM,而本研究中最强效抑制剂的Ki值在200nM左右;单独靶向μ-阿片受体时,最高Ki值为2.3μM³⁷,而本研究中最高Ki值为725nM。然而,此前的筛选仅涉及300万个分子,文库规模比本研究小几个数量级。μ-阿片受体/血清素转运体双靶点对接筛选中,仅有一个对接产物具有弱激动活性。
我们最终旨在寻找具有联合活性的配体,因此双靶点命中率低于单个靶点对接筛选。在α₂ₐ/血清素转运体筛选中,41种对接优先级化合物与α₂ₐ肾上腺素能受体结合(命中率39%),21种与血清素转运体结合(命中率20%),但仅16种与两个靶点均结合(双靶点命中率15%)。在μ-阿片受体/血清素转运体多靶点筛选中,26种化合物与血清素转运体结合(命中率47%),16种与μ-阿片受体结合(命中率29%),但仅6种与两个靶点均结合(双靶点命中率12%)。在μ-阿片受体/α₂ₐ肾上腺素能受体筛选中,未发现任何双靶点作用配体,这可能与两个靶点的低命中率有关(因此未进一步推进该组化合物)。α₂ₐ/血清素转运体的双靶点结合率是单个靶点命中率简单乘积的2倍,尽管其统计显著性尚不确定。更明确的是,双靶点结合率远高于将血清素转运体单靶点对接筛选产物用于α₂ₐ肾上腺素能受体活性测试³⁶,或将α₂ₐ肾上腺素能受体单靶点对接筛选产物用于血清素转运体活性测试⁴⁸——这两种情况下,浓度高达10μM 时均未检测到活性。
多靶点药理学的最终标准是功能,本研究旨在寻找α₂ₐ肾上腺素能受体和μ-阿片受体激动剂。在16种与α₂ₐ/血清素转运体均具有联合活性的配体中,5种为α₂ₐ肾上腺素能受体激动剂(表2);在6种与μ-阿片受体/血清素转运体均具有联合活性的配体中,仅1种为μ-阿片受体激动剂(表2)。α₂ₐ肾上腺素能受体激动剂的半数有效浓度(EC₅₀)范围为390nM-2.2μM,μ-阿片受体激动剂活性较弱。由于仅发现1种μ-阿片受体激动剂,且此前发现非激动剂可通过优化和立体化学纯化展现激动活性³⁸,因此我们将所有6种μ-阿片受体/血清素转运体双靶点活性配体均推进至下一步研究。总之,经过以下连续约束筛选:(1)单个靶点配体浓度<10μM;(2)双靶点结合;(3)受体激动活性,尽管每个步骤的成功率均较为可观,但在241种测试化合物中,最终仅获得5种α₂ₐ/血清素转运体多靶点配体和1种μ-阿片受体/血清素转运体多靶点配体。加上从μ-阿片受体/血清素转运体筛选中获得的5种非激动剂,共得到11种待推进的多靶点配体。
对接产物的优化
为优化初始筛选产物,我们首先寻找配体上不与任何一个靶点发生冲突的修饰位点,并进行配体建模(图4A-D)。由于血清素转运体(转运蛋白)与α₂ₐ肾上腺素能受体、μ-阿片受体(均为G蛋白偶联受体)的拓扑结构不同,对接配体上仅有少数此类位点。例如,化合物Z8779877149(z7149)深埋于α₂ₐ肾上腺素能受体的口袋中(图4A),限制了分子扩展;而血清素转运体的结合腔大得多,允许在更多位点进行修饰(图4B),但综合两个靶点的约束条件后,仅剩下一个可容纳修饰的位点(图4A,B)。第二种方法是不考虑受体结构,先对配体进行小型系统性修饰,然后利用Arthor和SmallWorld程序(https://sw.docking.org/,NextMove Software,英国剑桥)在含200亿个定制合成分子的文库中搜索,为11种母核化合物找到11,018种可合成的类似物。通过将这些类似物对接至成对靶点,筛选结合效果良好的分子并优先进行测试。通过以上两种方法,在第一轮优化中为11种母核化合物合成了171种类似物(补充信息文件verified-compounds.xlsx)。
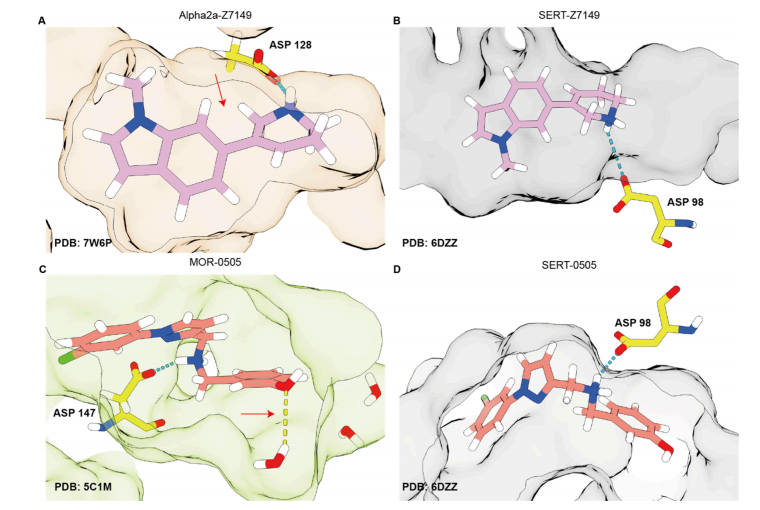
针对α₂ₐ/血清素转运体靶点对,我们聚焦于两种不同化学类型中活性最强的5种配体(表3)。对于第一种化学类型,我们发现α₂ₐ肾上腺素能受体活性提升6倍的类似物(化合物Z4387158014);对于第二种化学类型,发现血清素转运体抑制活性提升高达150倍的分子(化合物Z8727393901),但这两种情况均以丧失另一个靶点的活性为代价(表3)。对于以z7149和ZINCgw000000A08w(A08w)为代表的第三种化学类型,仅发现能维持双靶点活性但未提升活性的类似物。最终,选取6种化合物(2种对接初始产物和4种类似物)进行药代动力学研究(补充信息表1)。对接初始产物z7149表现出最佳的靶点活性和中枢神经系统暴露量。
z7149属于四氢吡啶类化合物,我们此前发现该类化合物对5-羟色胺₂ₐ受体具有活性且具有抗抑郁样作用³⁸。由于5-羟色胺₂ₐ受体与镇痛作用相关⁶¹,⁶²,且血清素转运体抑制剂是已知的抗抑郁药,因此我们测试了z7149对5-羟色胺₂ₐ受体的活性。令人鼓舞的是,z7149对5-羟色胺₂ₐ受体的活性与对α₂ₐ肾上腺素能受体和血清素转运体的活性处于同一浓度范围,基于Gq钙实验的激动剂EC₅₀为172nM,最大效应(Emax)为76%。这些特征与我们在四氢吡啶-5-羟色胺₂ₐ受体研究中观察到的结果相近,该研究已得到具有高效和长效抗抑郁样作用的化合物³⁸。z7149对α₂ₐ肾上腺素能受体、血清素转运体和5-羟色胺₂ₐ受体的靶点效价,以及相对良好的中枢神经系统暴露量,促使我们在镇痛、抗抑郁和抗焦虑类药物实验中对其进行测试(下文详述)。
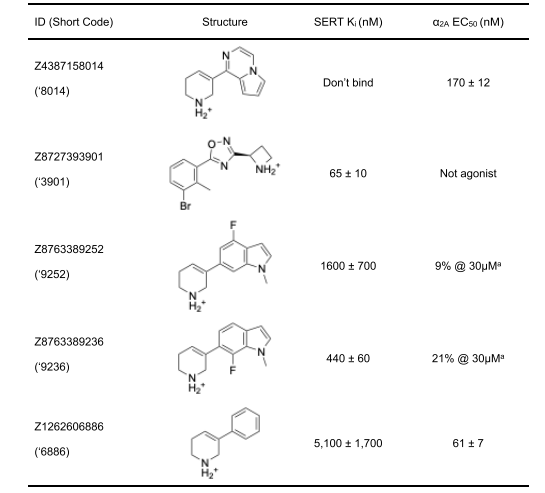
针对μ-阿片受体/血清素转运体靶点对,我们聚焦于6种活性最强的筛选产物,代表5种不同化学类型(表2)。μ-阿片受体的结合腔较大(图4C,D),允许更多衍生化修饰。在修饰过程中,我们将N-甲基苯胺改为苯酚,以改善与μ-阿片受体正构位点中有序水分子的氢键结合(图4C和表4),得到化合物Z8962410505(0505)。令人鼓舞的是,这一修饰将拮抗剂转化为激动剂,同时使血清素转运体活性提升6倍。后续修饰未进一步提高活性,因此将化合物0505推进至药代动力学研究(表4)。
综合α₂ₐ/血清素转运体和μ-阿片受体/血清素转运体靶点对的优化结果,我们为11种对接筛选产物合成了254种类似物。尽管成对靶点中单个靶点的配体亲和力相较于母核化合物可提升4-150倍,但对一个靶点的活性提升通常以另一个靶点的活性丧失为代价(表3)。最佳案例是μ-阿片受体/血清素转运体靶点对,将甲基苯胺替换为羟基后,两个靶点的亲和力均提升约6倍(化合物0505),血清素转运体的Ki值为250nM,μ-阿片受体激动剂的EC₅₀为140nM。即便如此,后续类似物合成仍未进一步改善双靶点活性(表4),且两组靶点对的配体活性均稳定在100nM级别。
脱靶效应
理想的多靶点化合物应对脱靶靶点具有选择性,而非通过非特异性实现多靶点作用。因此,我们评估了化合物对与血清素转运体密切相关的转运体(多巴胺转运体和去甲肾上腺素转运体)的抑制作用。z7149对多巴胺转运体和去甲肾上腺素转运体的Ki值分别为6.3μM和881nM,0505对二者的Ki值分别为922nM和3.2μM(补充信息图8)。采用Tango实验在10μM浓度下测试了两种化合物对320个人源G蛋白偶联受体的激动活性。0505对所有靶点均未表现出明显活性,而z7149仅对5-羟色胺₂B受体(EC₅₀=15nM,Emax=34%)和5-羟色胺₂C受体(EC₅₀=26nM,Emax=19%)表现出显著活性。从治疗角度来看,z7149对5-羟色胺₂B受体的效价令人担忧,因为该受体与瓣膜病变相关⁶³,⁶⁴,但较低的效应强度在一定程度上缓解了这一风险。总体而言,这些分子在上述实验中未表现出广泛的非特异性。
冷冻电镜结构测定
为指导后续化合物优化、从原子层面理解活性机制并验证对接预测结果,我们采用单颗粒冷冻电镜测定了z7149与α₂ₐ肾上腺素能受体、z7149与血清素转运体、0505与μ-阿片受体的结合结构(方法部分)。结构的整体标称分辨率为3.29-3.40Å,受体和转运体正构位点的分辨率为3.3-3.6Å。冷冻电镜图谱显示,跨膜区域密度清晰连续,可明确构建正构位点和结合配体的模型。
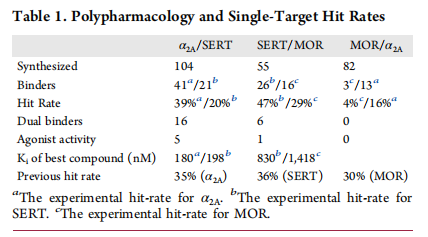
z7149与α₂ₐ肾上腺素能受体结合的冷冻电镜模型与对接构象的配体重原子均方根偏差(RMSD)为1.22Å(图4A),与预测一致,与天冬氨酸128形成离子对。在z7149与血清素转运体结合的冷冻电镜结构中,受体呈现闭合-闭塞构象,跨膜螺旋1(残基85-95)摆动至正构位点。此前的对接筛选基于血清素转运体的内向开放构象³⁶。尽管受体发生构象变化,但对接预测的构象与实验模型中的相互作用残基和结合位点一致(图5C);部分由于构象变化,对接构象与实验构象的配体均方根偏差升至2.97Å。然而,若选择z7149的高对接构象,配体均方根偏差可降至1.91Å。与对接模型(图4B)相比,闭塞构象的腔更小(图5D),这与Z8763389252等较大类似物的效价降低一致(表2)。比较z7149与α₂ₐ肾上腺素能受体和血清素转运体的复合物发现,四氢吡啶环发生旋转,使其阳离子氮原子处于不同相对位置,分别与α₂ₐ肾上腺素能受体的天冬氨酸128或血清素转运体的天冬氨酸98形成氢键,这种能力可能有助于非序列相关靶点之间的多靶点作用。
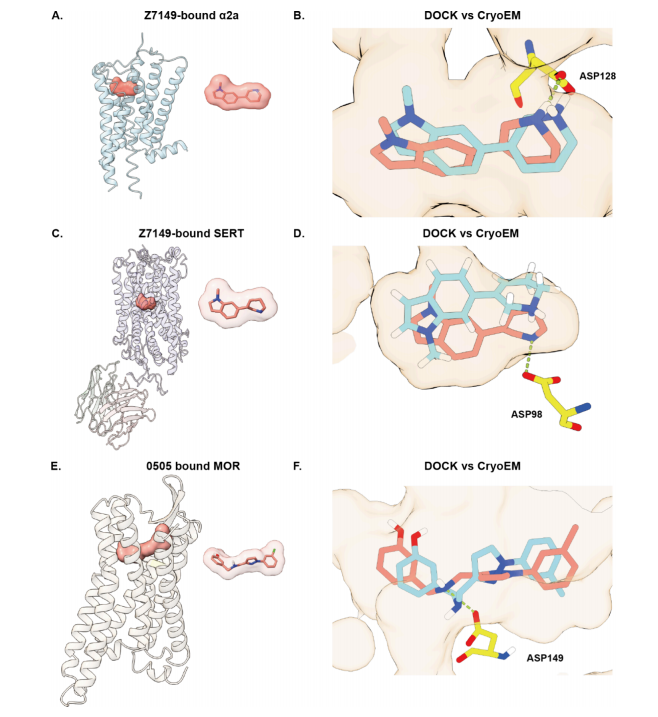
对于μ-阿片受体/0505复合物,配体的冷冻电镜图谱也基本支持对接构象。在实验复合物中,0505与天冬氨酸149和有序水分子形成氢键(与预测一致),该位点通常为受体的苯酚识别位点。实验构象与对接构象的均方根偏差为2.1Å(图5E)。
多靶点化合物的体内活性
尽管z7149和0505的效价比体内活性探针通常使用的效价低10-30倍,但其多靶点作用可能相较于单靶点药物产生新的或增强的活性。因此,我们首先探究了它们的药代动力学特征,评估化合物在大脑中的暴露情况。以10mg/kg剂量腹腔注射后,两种化合物均具有显著的中枢神经系统暴露量,z7149的血药峰浓度(Cmax)为79.7μMol,0505为14.8μMol,半衰期分别为56分钟和117分钟(补充信息图9-21和表1)。然而,作为大脑游离分数替代指标的脑脊液暴露量较低⁶⁵,⁶⁶,z7149的脑脊液浓度为830nM,0505仅为27nM。这一结果表明,z7149在血药峰浓度时对α₂ₐ肾上腺素能受体、血清素转运体和5-羟色胺₂ₐ受体的半数有效浓度覆盖率为2-4倍,而0505对μ-阿片受体和血清素转运体活性的覆盖率不足0.3倍。这一覆盖率水平使得0505难以产生体内活性,事实上在初步镇痛实验中,该化合物未表现出明显活性(补充信息图9)。因此,未进一步推进0505的研究。
z7149的药代动力学特征更为理想,以10mg/kg剂量腹腔注射后,在行为学实验中观察到其镇痛和抗异常性疼痛活性。测试包括:甩尾实验(50℃)和哈格里夫斯实验(评估对伤害性刺激的反射反应)、热板实验(评估对伤害性热刺激的反射和情感反应,如舔爪)、神经性疼痛的spared神经损伤模型以及炎症性疼痛的完全弗氏佐剂模型(图6)。令人鼓舞的是,与其他对接得到的α₂ₐ肾上腺素能受体激动剂(如9087)类似,z7149在转棒实验中未表现出镇静作用,表明其镇痛作用并非源于运动障碍或镇静。与9087不同,z7149未诱导条件性位置偏爱。这种差异可能反映了z7149对血清素转运体的多靶点作用——血清素转运体抑制可抑制条件性位置偏爱⁶⁷,⁶⁸,而9087不具备这一活性⁴⁸。需注意的是,我们尚未证实二者的因果关系,血清素转运体在z7149不诱导条件性位置偏爱中的作用目前仍为推测(图6)。
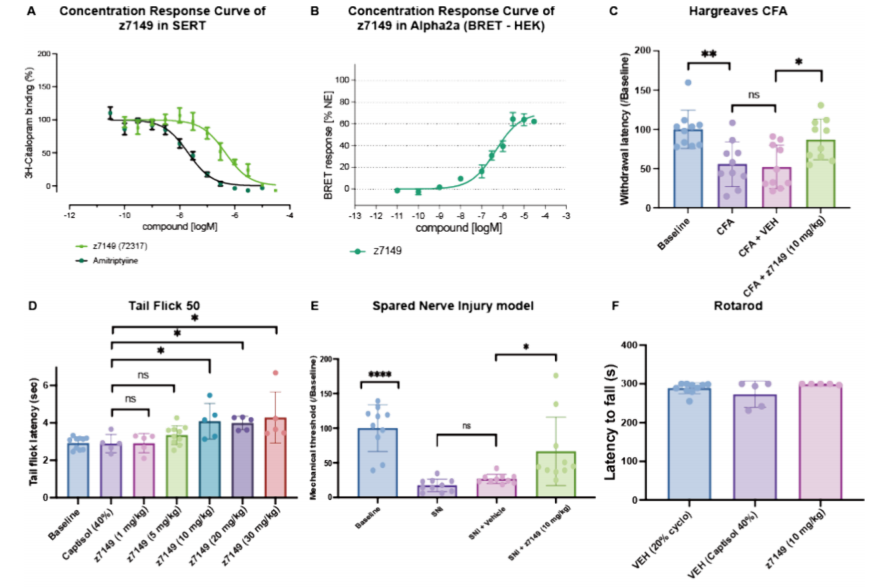
由于z7149同时具有5-羟色胺₂ₐ受体和血清素转运体活性,我们还在小鼠中测试了其抗焦虑和抗抑郁类药物活性(图7A-C)。在通过足底电击评估惩罚性穿越的四板实验中,腹腔注射0.25mg/kg z7149的小鼠穿越次数几乎是溶媒处理组的两倍,其抗焦虑样效果与腹腔注射0.5mg/kg阳性对照药地西泮相近。同时,在对囊泡单胺转运体2杂合小鼠(易表现出抑郁样表型)⁶⁹的悬尾实验中,0.2和0.5mg/kg剂量的z7149具有抗抑郁样作用,持续时间长达5天。该作用的强度和持久性均优于氟西汀:氟西汀的剂量是z7149的100倍,但其急性疗效最多与该新型多靶点配体相当(图7C,30分钟时间点),且氟西汀的疗效随时间下降,到第3天完全丧失。尽管四氢吡啶类5-羟色胺₂ₐ受体激动剂的长效作用已有先例³⁸,但考虑到z7149的药代动力学覆盖率较低(半数有效浓度/血药峰浓度),其作用效价可能反映了5-羟色胺₂ₐ受体和血清素转运体的双重激活——早期系列化合物未显著抑制血清素转运体。类似地,该分子的抗焦虑样作用也可能源于其多靶点特性。需注意的是,剂量响应显示,当剂量升至1mg/kg时,疗效下降,这可能反映了z7149通过α₂ₐ肾上腺素能受体激动作用产生的多靶点负效应,因为α₂ₐ肾上腺素能受体拮抗被认为具有抗抑郁作用⁷⁰。同样,5-羟色胺₂ₐ受体激动/血清素转运体抑制的联合作用在体内疗效中的机制尚未明确证实。
4、讨论
大型文库多靶点对接技术成功筛选出具有双靶点和三靶点联合活性的分子。四项关键发现值得强调:首先,随着对接文库规模扩大,潜在的多靶点化合物会自然出现(图1和补充信息图1),且在回顾性双靶点对接中,这些化合物在两个靶点中均排名靠前(补充信息图2-4);其次,在针对具有生物学相关性但结构不同靶点的前瞻性双靶点对接中,发现了多种可结合两个靶点的配体。尽管这些分子靶点对的三级结构不同,但其结合位点仍具有相似性且尺寸相近。相反,μ-阿片受体/α₂ₐ肾上腺素能受体的正构位点尺寸存在差异,未发现大量双靶点结合配体,表明对接技术无法克服靶点之间固有的物理化学性质不匹配;第三,即使对于发现多个筛选产物的靶点对,基于结构的优化也比单个靶点筛选的优化更具挑战性³⁶,³⁷,⁴⁸。尽管类似物合成可使单个靶点的效价提升高达150倍,但很少有分子能同时改善两个靶点的活性,更常见的是一个靶点活性提升而另一个靶点活性丧失;第四,尽管联合活性相对温和,但至少有一种分子(z7149)在小鼠实验中具有抗异常性疼痛活性,并具备抗焦虑和抗抑郁类药物作用。其不诱导条件性位置偏爱的特性使其区别于9087等早期单靶点筛选产物,这可能源于对血清素转运体的新型激活作用。类似地,z7149强效的抗抑郁和抗焦虑类药物活性可能源于血清素转运体激活与5-羟色胺₂ₐ受体活性的联合作用。
本研究存在一定局限性:z7149和0505的效价均低于早期研究中的单靶点激动剂³⁷,⁴⁸。这部分源于对接筛选产物的效价较低,更重要的是多参数优化(此处为同时针对两个靶点进行优化)的挑战。类似地,化合物的药代动力学特征也不如早期单靶点分子,这可能也反映了多靶点优化的约束条件,以及配体优先级排序流程中缺乏早期药代动力学性质计算——这是当前的研究热点。生成式人工智能⁷¹,⁷² 或机器学习⁷³⁻⁷⁵等方法能够在超大空间中进行引导搜索,具有应用前景,但前瞻性测试仍较少。与血清素转运体的单靶点筛选类似,尽管对接配体占据了正确的位点并形成了实验结构中观察到的大部分相互作用,但转运体的整体构象从对接所基于的内向开放结构发生了变化。同样,包括柔性受体对接在内的更先进方法具有应用前景,但在大型文库筛选中引入诱饵结构仍是一项挑战⁷⁶⁻⁷⁸。最后,由于许多强效分子仍相对较小,无法排除多种结合模式的可能性,尽管这些靶点共有的离子相互作用可能降低了这种可能性。
总体而言,研究结果喜忧参半。对于结合位点大致相似但折叠方式不同的靶点对,大型文库对接技术能够发现在两个靶点中均排名靠前的分子,且单个靶点的命中率相对较高。当要求配体对两个靶点均具有活性时,命中率会下降,且双靶点活性配体的活性仅处于100nM-低微摩尔范围,弱于针对相同靶点的单蛋白筛选中最强效的分子³⁶,³⁷,⁴⁸。此外,我们在双靶点活性产物的联合优化中仅取得了有限成功,仅有一种分子(z7149)展现出体内疗效。该化合物确实表现出可能源于多靶点作用的新治疗活性:z7149在急性和慢性疼痛模型中均有效,且未诱导早期缺乏血清素转运体活性的激动剂所特有的条件性位置偏爱。尽管z7149在中枢神经系统中的效价-暴露比相对较低,但仍具有强效的抗抑郁和抗焦虑类药物活性,这可能反映了其对血清素转运体和5-羟色胺₂ₐ受体的联合活性。因此,本研究表明,尽管双受体靶向的约束条件会降低配体发现和优化各阶段的结果,但文库对接技术仍可用于治疗多靶点药理学具有优势的疾病,即使靶点不属于同一家族。这些不足可能通过进一步扩大对接可及文库的规模(目前已达到万亿级)来克服。相反,对于双靶点激活具有优势但折叠方式不同的蛋白质,多药联用方法也可能值得研究。
5、材料与方法
分子对接
对接参数与此前的单靶点研究保持一致,对接分子来自ZINC22数据库的定制合成分子:针对α₂ₐ/血清素转运体和α₂ₐ/μ-阿片受体靶点对,对接3300万个化合物;针对μ-阿片受体/血清素转运体靶点对,对接9亿个化合物。采用基于ECFP4的塔尼莫托系数(Tc<0.35)对每个靶点的已知配体进行新颖性筛选,筛选出排名前300,000的共享分子。利用相互作用指纹筛选去除几何结构上缺失关键相互作用或暴露未补偿氢键供体的化合物,重点关注血清素转运体的天冬氨酸98、α₂ₐ肾上腺素能受体的天冬氨酸128和μ-阿片受体的天冬氨酸149之间的氢键。设置应变筛选阈值>2kcal/mol,去除高内能构象的分子。采用基于ECFP4的塔尼莫托系数(Tc=0.5)对剩余分子进行拓扑聚类,得到约1000个高排名聚类代表物,通过视觉inspection评估其在两个结合口袋中的结合情况。优先选择与两个靶点均具有良好相互作用的化合物进行从头合成和测试。
化合物合成与纯度
所有测试化合物均由Enamine公司合成和纯化(详见补充信息中的合成和纯化部分)。所有化合物通过液相色谱-质谱联用仪检测的纯度均不低于90%,活性化合物通过核磁共振氢谱进一步确认。活性化合物的纯度通过液相色谱-质谱联用仪和核磁共振氢谱检测为90%-95%以上,其中248种对任意靶点具有活性的分子中,196种纯度为95%或更高;71种对两个或多个靶点具有联合活性(每个靶点≤10μM)的分子中,59种纯度为95%或更高。所有推进至药代动力学和体内行为学研究的化合物纯度均不低于98%。
配体优化
在第一轮类似物合成中,以初始对接筛选产物(图3)为查询对象,利用SmallWorld(https://sw.docking.org/)和Arthor(http://arthor.docking.org)程序(NextMove Software,英国剑桥)在含200亿个分子的Enamine REAL文库中搜索,收集与母核化合物最多相差3个重原子的分子。如前所述,将这些分子对接至靶点以评估互补性,优先选择结合效果良好的类似物进行测试。针对所有三个靶点,根据第一轮观察到的亲和力变化,进一步选择两轮类似物,偶尔用于测试特定的相互作用假设。