提起Phil S. Baran教授,在有机化学界堪称殿堂级标杆,其学术影响力无人不晓。今天要为大家重磅分享一篇突破性力作——由Enamine公司首席科学家Pavel K. Mykhailiuk博士与Baran教授联合担任通讯作者的研究论文。该工作跳出传统合成框架,以二溴卡宾与双环[1.1.0]丁烷的加成反应为核心,开创性地实现了无需依赖[1.1.1]丙二烯的取代双环[1.1.1]戊烷(BCPs)规模化合成,彻底破解了长期困扰领域的“丙二烯难题”,为药物化学中这一关键生物电子等排体的广泛应用开辟了全新路径。
二溴卡宾与双环 [1.1.0] 丁烷的加成反应:一种合成取代双环 [1.1.1] 戊烷的简便方法
含张力的多环烃类化合物已成为药物研发中日益重要的结构单元。特别是取代双环[1.1.1]戊烷(BCPs),作为广泛存在的苯环的生物电子等排体,其地位日益凸显。尽管BCPs具有优良的药代动力学性质,但现有合成策略存在显著缺陷——主要是过度依赖[1.1.1]丙二烯这一原料。该原料操作难度大,不仅使规模化生产变得复杂,还阻碍了这类结构单元的广泛应用。本研究报道了2,2-二溴双环[1.1.1]戊烷的合成方法,提供了一类用途广泛的取代BCPs,且无需依赖基于[1.1.1]丙二烯的前驱体。我们通过简单廉价的工艺实现了这类化合物的规模化制备,并通过合成多种高价值的合成砌块(包括备受关注的桥连杂芳基化BCP衍生物,其通过电催化交叉偶联反应制备),凸显了它们在药物化学研究中的应用价值。
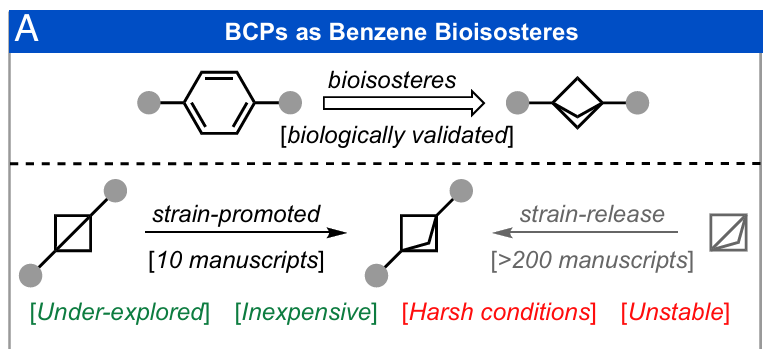
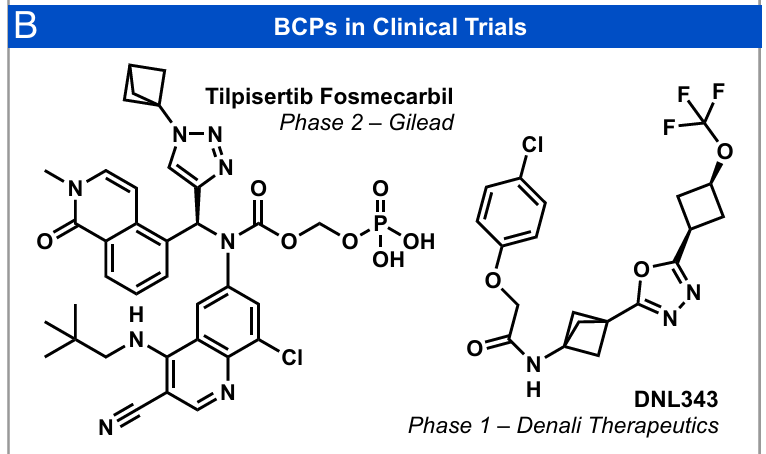
取代苯环是生物活性分子中普遍存在的结构(1)。然而,药物化学领域的最新趋势强调,需要通过逃离平面结构“Escape from Flatland”(2, 3),用饱和度更高、三维结构更丰富的替代骨架取代平面芳环(4, 5)。双环[1.1.1]戊烷(BCPs)已成为理想的生物电子等排体(图1A),相比其苯环类似物,通常表现出更优的溶解性、代谢稳定性和结合特异性(6-10)。事实上,Gilead和Denali Therapeutics开发的含BCP骨架的候选药物已进入临床试验阶段(图1B)(11, 12),这凸显了该结构单元在药物化学研究中的重要性。
目前获取BCP片段最常用的策略(13)均以高张力烃[1.1.1]丙二烯为起始原料(14)。通过自由基或极性路径打开[1.1.1]丙二烯的环,可得到有价值的桥头取代BCPs(15-18)。尽管实现该过程的方法多种多样,但这类策略存在所谓的“丙二烯难题”(13)。具体而言,使用 [1.1.1]丙二烯存在诸多实际问题,即成本较高、反应条件苛刻、母体烃相对不稳定,且在溶液中处理这种挥发性化合物时操作难度较大(13)。这些因素限制了基于丙二烯的方法的实际应用,因此亟需开发全新的BCP骨架合成路径(19-21),尤其要聚焦适用于工业规模化应用的方法,以加速BCP在治疗药物研发中的应用。
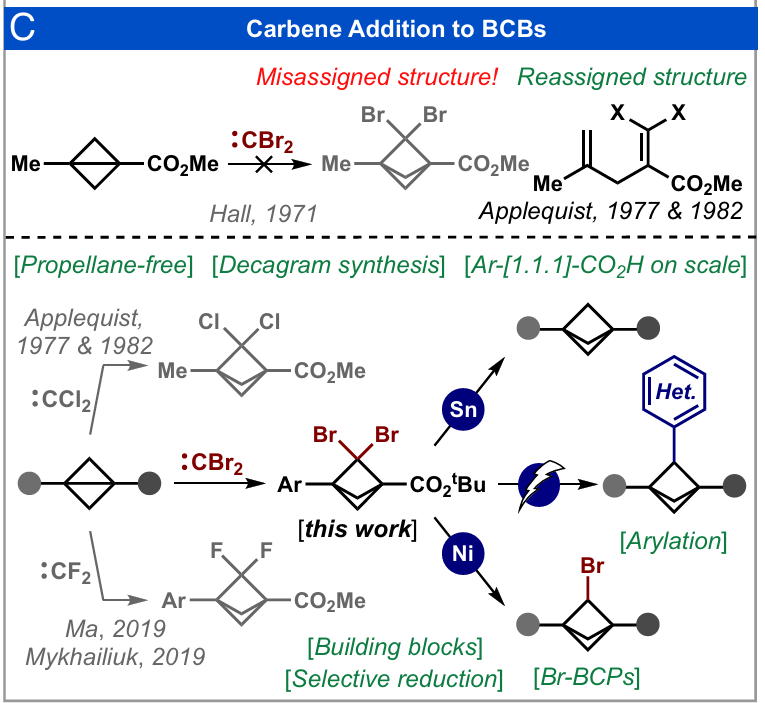
另一种较少使用的BCP合成路径是通过卡宾与双环[1.1.0]丁烷(BCBs)的加成反应(图1C)。已有报道称,二卤卡宾(:CX₂)(22-26)以及近年来发现的含多种碳基取代基的卡宾(27, 28),可用于合成结构多样的BCP产物。与[1.1.1]丙二烯相比,BCBs具有操作简便的显著优势,且可通过多种取代模式便捷制备,还能实现规模化生产,因此该合成路径成为一种极具吸引力的替代方案(29-31)。1971年,Hall等人报道了通过BCBs与相应二卤卡宾反应,看似成功制备了二氯BCP和二溴BCP,并指出两种卤代产物的光谱数据存在相似性(32)。1977年,Applequist通过对比合成法,明确将二氯BCP的结构修正为偕二卤烯烃(23),这一发现也对之前报道的二溴BCP的结构表征提出了质疑。随后,Applequist成功实现了二氯卡宾(:CCl₂)与BCBs的加成反应,制备出二氯BCP(22, 23)。1985年,Wiberg利用该产物合成了全氘代[1.1.1] 丙二烯(33),2016年,Hirst又通过该产物实现了治疗先导化合物的克级合成(6)。2019年,首次报道了通过二氟卡宾(:CF₂)合成桥连氟代BCP的方法(24, 25),后续研究进一步报道了生成:CF₂的其他条件以及这些合成砌块的衍生化反应(34-39)。2022年,我们报道了溴氟卡宾(:CBrF)与BCBs的加成反应,通过还原C-Br键,首次制备出可用于药物化学研究的桥连单氟代BCP(26)。
自1971年首次报道以来(32)的50多年间,据我们所知,尚未有关于更重的卡宾同系物(如二溴卡宾:CBr₂)与BCBs成功加成的后续报道(40)。尽管如此,桥头溴代的BCPs近年来受到广泛关注,它们可作为合成2-取代BCP骨架的有效前驱体,而2-取代BCP作为邻位或间位取代苯环的生物电子等排体具有重要价值(41, 42)。目前,已知的溴代BCP合成路径仅有一条(41),其起始原料最终仍源于[1.1.1]丙二烯。因此,上述“丙二烯难题”使该方法的普适性受到限制,亟需开发不依赖[1.1.1]丙二烯的取代BCP骨架新合成方法。
基于上述因素,我们致力于研究:CBr₂与BCBs的反应活性,以获取一类此前未被探索的二卤代BCPs。本研究报道了一系列结构多样的二溴BCPs的合成,并通过偕二溴单元的规模化选择性还原反应,制备出单溴BCP或双脱卤BCP(这类化合物均为药物化学家和工艺化学家的研究热点),同时还证明了二溴BCPs可作为合成多种结构多样BCPs的前驱体。此外,受近期利用单溴BCP骨架实现交叉亲电偶联(XEC)的研究启发(41),我们报道了一种镍电催化C(sp³)-C(sp²)交叉偶联反应,用于二溴BCPs的还原芳基化,可直接制备高价值的C2-修饰BCPs。
结果与讨论
二溴卡宾与 BCBs 加成反应的开发
我们以桥头含芳基和酯基的模型BCP(1a)为研究起点(图2)(26)。在获得合适的前驱体后,我们首先尝试将此前报道的溴氟卡宾与BCBs的反应条件(通过在相转移催化剂苄基三乙基氯化铵(BnEt₃NCl)存在下,用浓NaOH在甲苯中脱质子化二溴氟甲烷生成溴氟卡宾)(26)应用于本研究。
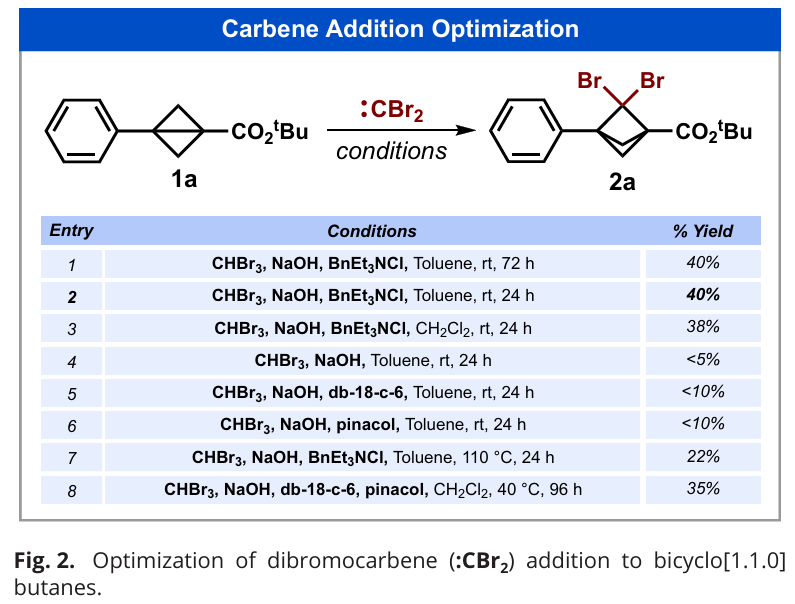
在本实验中,用溴仿替代氟利昂原位生成:CBr₂,成功以40%的可接受收率得到目标产物2a(实验序号1)(同时生成四卤代副产物,详见补充信息)。随后对其他反应条件的筛选表明,反应时间可缩短至24小时且收率不受影响(实验序号2),二氯甲烷与甲苯作为溶剂的效果相当(实验序号3)。若不添加相转移催化剂,仅能生成微量产物(实验序号4),且其他相转移催化剂的效果均不理想(实验序号5和6)。升高反应温度会导致收率下降(实验序号7),且相转移催化剂、溶剂和温度的其他组合也未能改善反应结果(实验序号8)(43)。
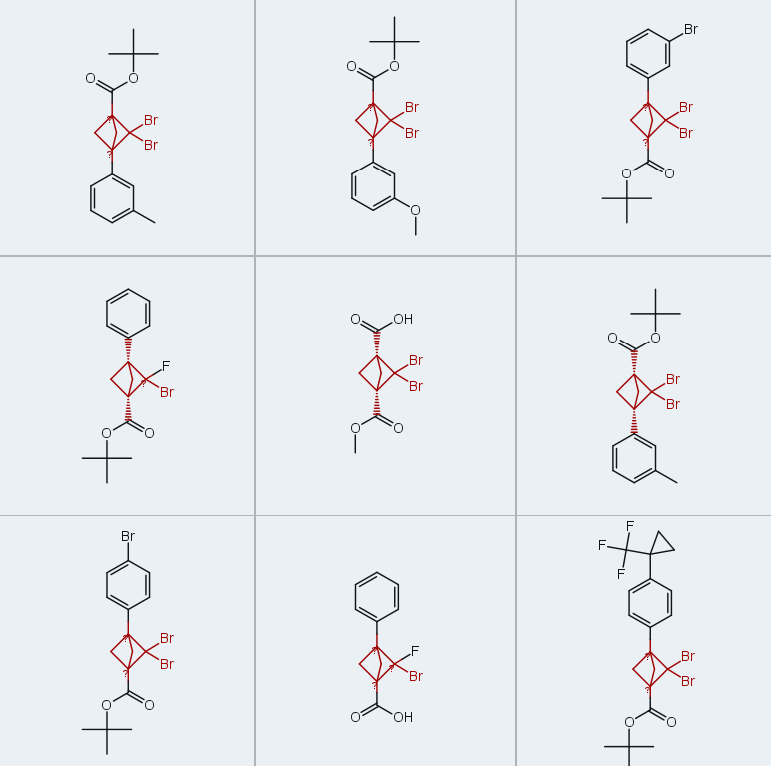
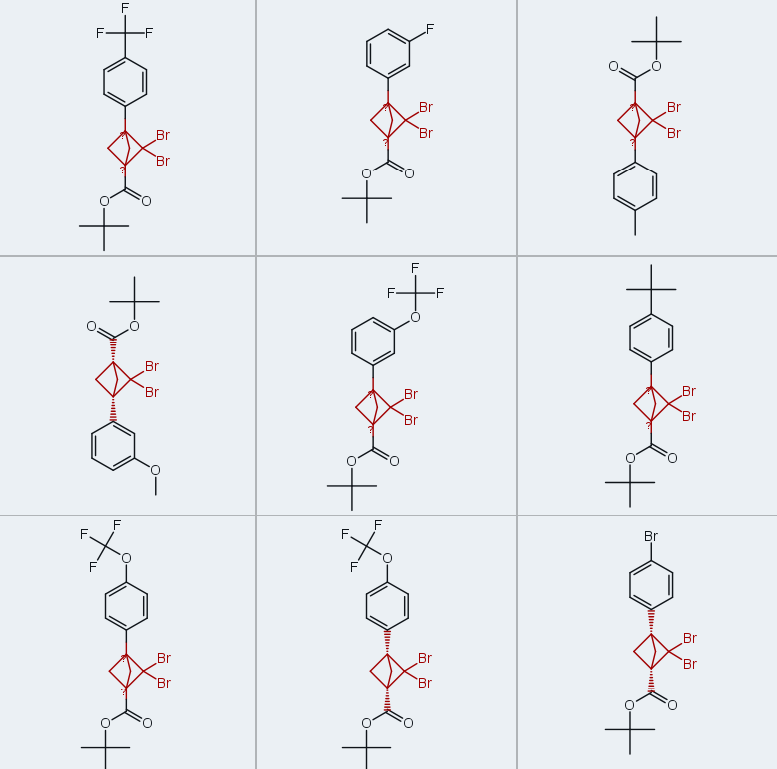
在确定了二溴卡宾与BCBs加成反应的有效方案后,我们合成了一系列在BCB 3-位具有不同芳基取代基的BCBs(1b-1o,四步反应收率为30%-53%)(图3),以验证卡宾加成反应的普适性。采用合成2a的最优反应条件(图2,实验序号2),成功合成了含简单给电子取代基(甲基:2b-2d、叔丁基:2e、甲氧基:2f;收率32%-41%)和吸电子取代基(氟:2g、2h、三氟甲基:2k、2l;收率29%-32%)的二溴BCPs,收率与母体化合物2a相近。芳基溴化物2i和2j(收率分别为34%和30%)为后续进一步官能团化提供了额外位点(37),而三氟甲基醚衍生物(2m:收率31%;2n:收率33%)和三氟甲基环丙基衍生物(2o:收率34%)在药物化学领域具有研究价值(44-46)。
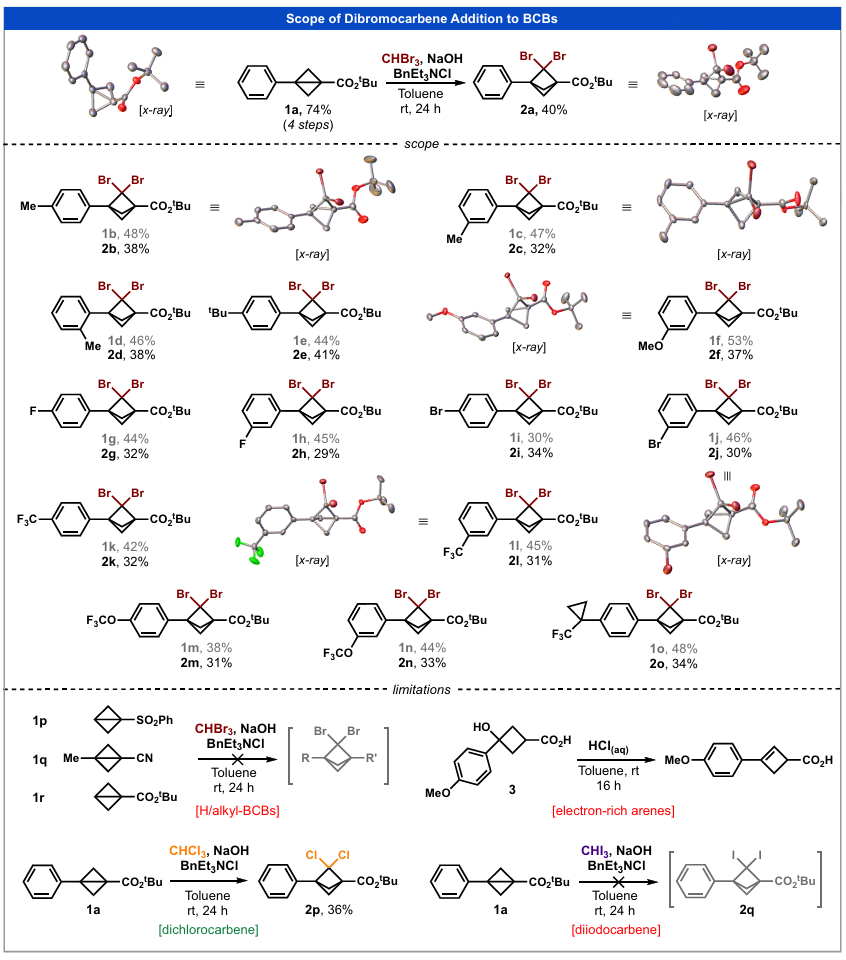
该反应存在一定局限性:不含芳基取代基的BCBs(1p-1r)无法参与卡宾加成反应;尽管可用氯仿替代卡宾前驱体制备二氯BCP(2p),但使用更重的碘仿无法得到目标二碘BCP(2q)。此外,BCB前驱体的制备也存在局限性,富电子芳烃(如化合物3)与该合成路径不兼容。
接下来,我们研究了卡宾加成反应的规模化可行性(图4)。根据文献报道的方法(26),前驱体BCB(1a)可由廉价易得的环丁烷衍生物(每克0.64美元)制备,且该过程无需柱层析分离,极大地提高了时间和成本效率。将卡宾加成反应规模扩大至75克时,2a的收率略有下降(35%)。叔丁酯脱保护后得到游离羧酸合成砌块8,通过该工艺单次可分离得到38克稳定的产物。
二溴 BCPs 的物理化学性质
在明确了二溴卡宾加成反应的适用范围和局限性后,我们进一步研究了二溴BCPs的物理化学性质(图5)。
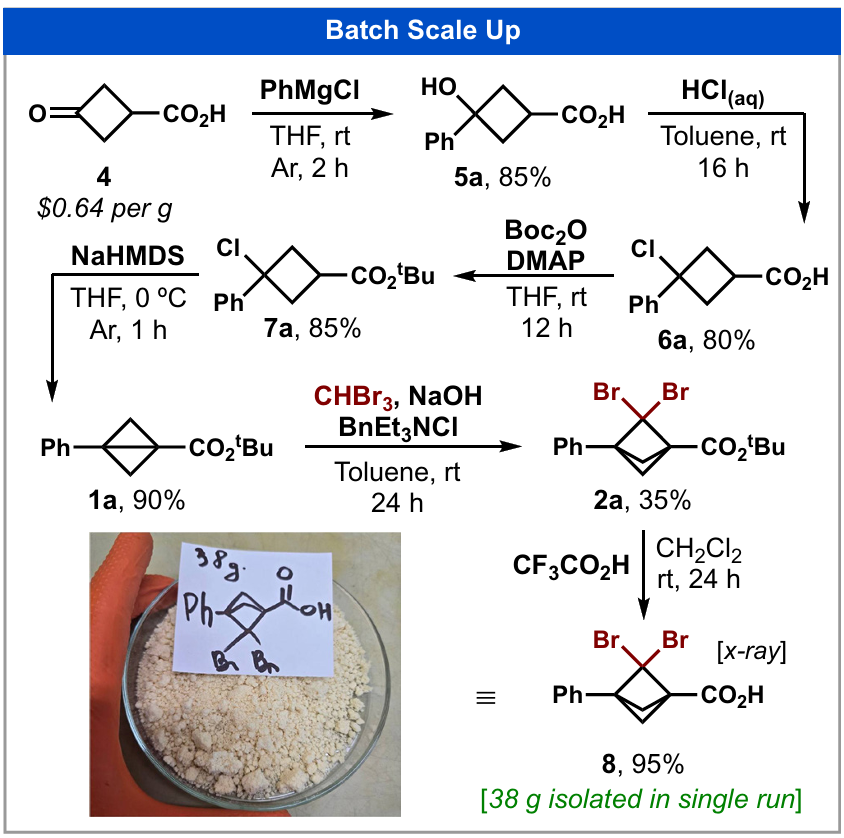
首先,我们通过实验测定了羧酸8(含CBr₂)和羧酸9(含 CH₂)的酸性。在BCP核心中引入偕二溴基团后,羧基的酸性降低了一个pKa单位:化合物9(CH₂)的pKa为5.1,而化合物8(CBr₂)的pKa为4.1。有趣的是,这种影响与偕二氯类似物11(pKa=4.1)相近,但弱于偕二氟类似物10(pKa=3.9)。
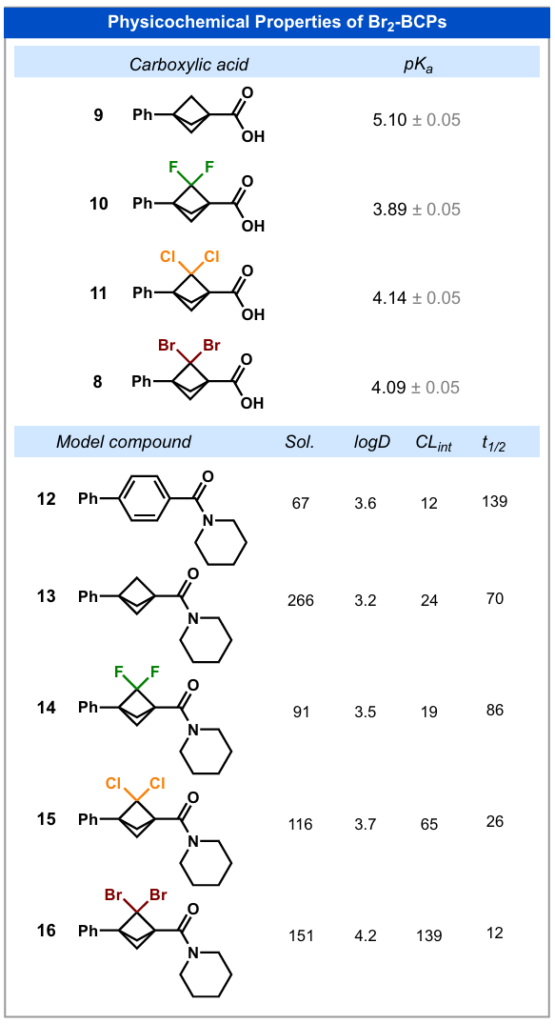
随后,我们合成了五种模型酰胺(化合物12-16),并研究了它们的溶解性、亲脂性和代谢稳定性。在BCP骨架中引入偕二溴基团后,水溶性降低(化合物13:266微摩尔/升vs化合物16:151微摩尔/升);亲脂性升高,logD值从3.2(化合物13)增至4.2(化合物16);代谢稳定性显著下降,内在清除率(CLint)从24微升・分钟⁻¹・毫克⁻¹(化合物13)增至139微升・分钟⁻¹・毫克⁻¹(化合物16)。这些结果表明,在药物化学研究中,二溴BCP不适合作为苯环或BCP环的替代物。相比之下,水溶性更强、稳定性更高且亲脂性更低的二氟BCP,在类似替代应用中更具潜力。尽管二溴BCPs存在代谢稳定性不足的问题,但通过C-Br键的官能团化,它们可作为制备取代BCPs的理想起始原料。
二溴 BCPs 的物理化学性质
接下来,我们研究了二溴BCPs的反应活性,以合成多种有用的合成砌块(图6)。
在十克级规模下,成功实现了二溴BCP(8)的桥头修饰,且收率良好(图6A)。羧基酯化反应得到甲酯17,随后苯环经氧化裂解生成单甲基二羧酸酯BCP(18),该化合物是一种有价值的正交保护合成砌块,可用于进一步衍生化反应。
考虑到无卤BCPs的广泛应用以及单溴BCPs的新兴研究价值,我们进一步探索了偕二溴官能团的还原条件,以获取这些备受关注的结构单元。
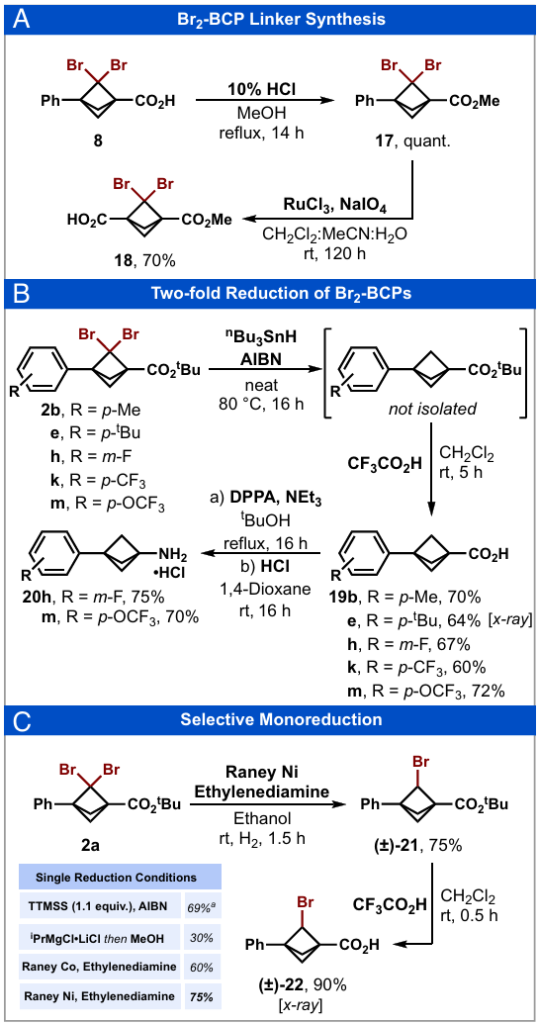
根据文献中关于二氯BCPs双还原反应的报道(6, 22, 33),我们将还原剂三丁基锡烷与自由基引发剂偶氮二异丁腈(AIBN)组合,应用于二溴BCPs(2b、2e、2h、2k和2m)的还原反应(图6B)。令人满意的是,所有化合物均成功被还原,粗反应混合物经三氟乙酸(TFA)处理后,可通过简单分离得到游离羧酸(19b、19e、19h、19k和19m),两步反应收率为60%-72%。随后,游离羧酸BCPs(19h和19m)经改进的库尔提乌斯重排反应,以良好收率(分别为75%和70%)得到BCP胺类化合物(20h和20m)。重要的是,这些转化反应可在十克级规模下进行,为获取高价值BCP片段提供了便捷途径。
在建立了二溴化物完全还原为相应BCP的条件后,我们进一步致力于开发选择性单还原方法,以拓展单溴BCPs的获取途径,用于BCP桥头的后期修饰(图6C)。对于二氯BCPs,已有文献报道使用三丁基锡烷或三(三甲基硅基)硅烷(TTMSS)与AIBN等自由基引发剂组合,可实现选择性还原(47, 48)。我们推测这些条件可用于二溴BCP(2a)的还原反应。尽管TTMSS具有一定效果,可通过核磁共振(NMR)测定得到目标BCP(±)-21的收率为69%,但目标化合物的分离过程较为繁琐,因为微量起始原料和完全脱溴的BCP会作为不可分离的副产物存在。随后,我们尝试通过Knochel的turbo-Grignard试剂进行金属-卤素交换(49),再通过Brønsted–Lowry酸淬灭反应。尽管该路径可使起始原料完全消耗,但目标单溴化物(±)-21的收率仅为30%。
在继续寻找合适的还原条件过程中,我们将注意力转向Raney metals。已有报道称,雷尼金属可用于二溴环丙烷的单还原反应,且选择性通常取决于取代基的空间位阻效应(50)。事实上,在我们之前的研究中,雷尼镍已被证明是溴氟BCP还原反应的高效催化剂(26)。在本研究中,二溴BCP(2a)与雷尼镍和乙二胺在氢气氛围下反应,可高选择性地得到目标BCP(±)-21,收率为75%。反应活性较低的雷尼钴也可用于该转化反应(50),但反应速率较慢且收率较低。(±)-21经TFA处理后,得到结晶固体游离羧酸(±)-22。
基于单溴BCPs的桥头官能团化
在建立了获取单溴代BCP(±)-21的可靠方法后,我们进一步制备了具有特定官能团的桥头衍生物,这些官能团已被证明适用于2-位的后期衍生化反应(图7)。近年来有研究表明,单溴BCP二酯可作为底物,通过金属光氧化还原催化实现BCP桥头的官能团化(41)。单溴BCP(±)-21经氧化裂解反应生成C3-羧酸,随后经酯化反应直接得到不对称二酯(±)-23(图7A)。在该化合物中,两个酯基的正交性使其能够实现每个桥头的逐步修饰,为文献中已报道的多种转化反应提供了模块化途径(51)。
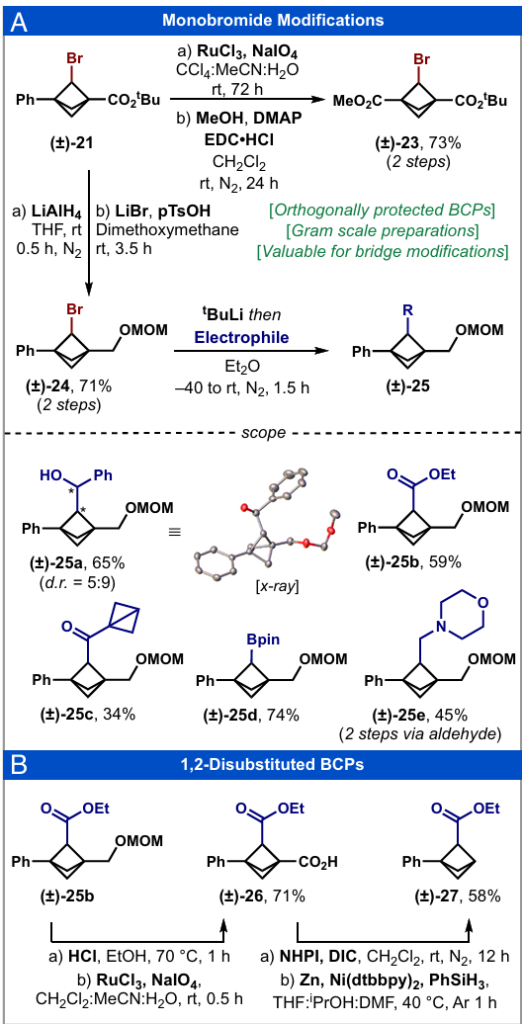
此外,已有报道称,桥头均含甲氧基甲基(MOM)醚取代基的单溴BCPs,可通过BCP烷基锂物种的配位稳定作用实现锂化反应,进而与亲电试剂发生捕获反应(42)。单溴BCP(±)-21中的叔丁酯经还原反应后,所得醇羟基经保护得到不对称单溴BCP(±)-24,该化合物仅含一个桥头MOM醚官能团,且可通过多克级规模制备。在改进的文献条件下(图7A)(42),不对称底物(±)-24经锂化反应后与亲电试剂捕获,成功得到含多种取代基(醇、酯、酮、频哪醇硼酸酯(Bpin)和胺)的衍生化产物(±)-25a-25e,收率为34%-74%。不对称单溴 BCP(±)-24与锂化反应的兼容性表明,仅需一个MOM螯合基团即可实现修饰反应。因此,可通过程序化正交策略,避免繁琐的官能团操作来实现桥头的去对称化,从而为后续官能团化提供更简洁的合成路径。事实上,2-位酯基衍生物(±)-25b经MOM脱保护和氧化反应,成功得到羧酸(±)-26(图7B)(52)。随后经脱羧反应得到1,2-二取代BCP(±)-27(53),该骨架作为邻位或间位取代苯环的生物电子等排体具有重要研究价值(4, 41, 52)。
二溴BCPs的电催化芳基化反应
烷基溴官能团在交叉亲电偶联(XEC)反应中应用广泛,尤其适用于C (sp³)-C (sp²) 键的构建(54)。镍电催化交叉偶联反应已被证明适用于规模化生产,且具有较高的官能团耐受性和化学选择性(55-57)。由于BCP C2-位芳基化的合成方法有限(19, 41, 52),我们推测具有还原不稳定性的溴代基团可作为该类衍生化反应的便捷位点。偕二溴BCP(2a)的还原电位显著高于单溴BCP(±)-21(循环伏安法数据详见补充信息),使其成为更适合XEC反应的前驱体。我们推测,电催化交叉偶联反应可提供一种简洁的C2-单官能团化路径,通过单一反应条件即可实现芳基化反应,随后剩余的C-Br 键被还原,无需额外的还原步骤。
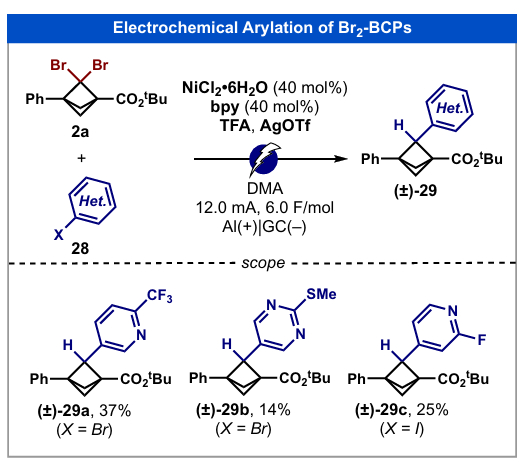
在当前已实现的反应体系中,二溴 BCP(2a)与杂芳基溴化物和碘化物(如 28)经镍电催化交叉偶联反应,得到双还原单芳基化BCPs(±)-29a-29c,收率为14%-37%(图8)。该偶联反应使用廉价的六水合氯化镍(NiCl₂・6H₂O)和联吡啶构建催化剂体系,并以三氟乙酸(TFA)和三氟甲磺酸银(AgOTf)作为添加剂。已有报道称,银盐和Brønsted酸是镍电催化交叉偶联反应的有效添加剂(57, 58)。对照实验表明,镍、配体、电能以及银盐和TFA均为该反应的关键添加剂(详见补充信息)。
结论
综上所述,本研究通过操作简便、可规模化的工艺,成功合成了一系列桥头官能化的二溴BCPs。这些化合物进一步拓展了BCP的化学空间,为药物化学家和工艺化学家提供了合成桥头取代BCPs的优良路径。重要的是,所报道的方案无需依赖[1.1.1]丙二烯作为关键合成前驱体。此外,本研究还证明,这些化合物可作为获取不对称单溴BCPs的重要原料,用于BCP桥头的后期修饰,同时为偕二溴BCPs作为电还原XEC反应的还原不稳定前驱体提供了概念验证。我们预计,这种易于规模化的工艺将进一步促进结构多样BCPs的获取,推动其在学术界和工业界的广泛应用。
值得关注的是,本文研究中涉及的关键中间体及系列高价值合成砌块,目前已在Enamine官网正式上线发售:
点此阅读原文了解更多。