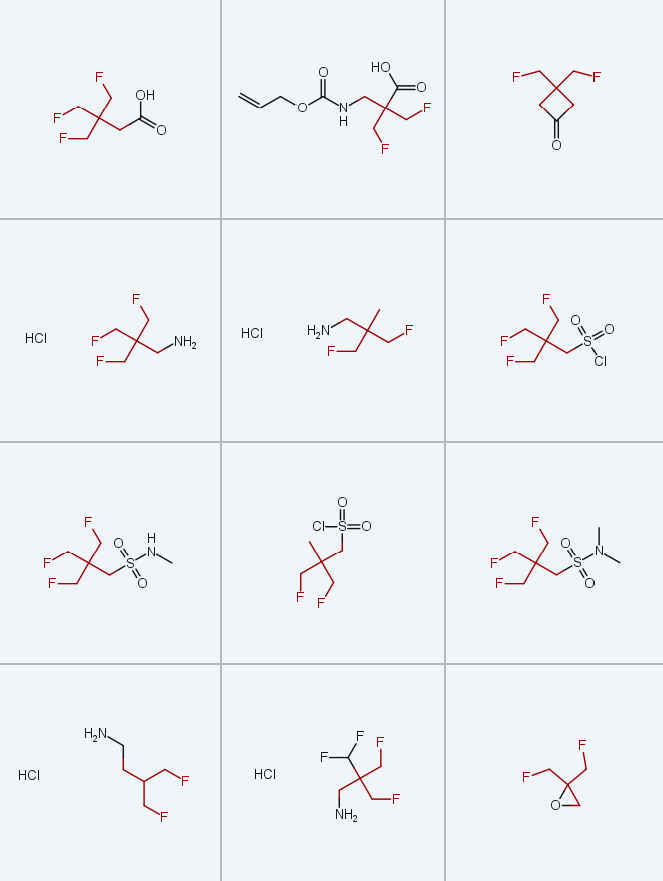
1、摘要
本文报道了以商用3,3-双(溴甲基)氧杂环丁烷为起始原料,简洁、可规模化合成三(氟甲基)烷基砌块(包括胺、羧酸、醇和溴化物)的方法。通过测定相应胺类和羧酸类化合物的解离常数(pKd)与脂水分配系数(Log P),并与低氟取代及无氟取代的同类物进行对比分析,完成了全面的理化特征表征。结果表明,三(氟甲基)甲基(β,β',β''-三氟叔丁基,(FCH2)3C,简称 (TFTB)基团虽空间位阻体积显著更大,但其电子效应和极性调节特性与三氟甲基(CF3)、2,2,2-三氟乙基(CF3CH2)取代基相似。构象分析显示,(FCH2)3C基团受空间静电相互作用调控,仅存在少量低能稳定构象,其中C=O⋅⋅⋅H−C和C−F⋅⋅⋅H−C相互作用对构象稳定起到关键作用。分子静电势分析阐明了该基团反常的脂溶性变化规律,以及其类三氟甲基的极性调节特征。上述研究数据均证实,TFTB基团是药物设计中极具价值的等排取代基序。
2、引言
在当代药物设计中,引入氟原子已成为核心策略之一,原因在于氟原子能以微小的结构扰动,调节小分子的电子、空间位阻及构象属性[1,2]。氟具有高电负性、小范德华半径,且碳-氟(C-F)键键能极高[3],因此引入氟原子通常能提升化合物的代谢稳定性[4]、调节酸碱性[5]、调控构象行为[6],同时不会显著改变分子尺寸。
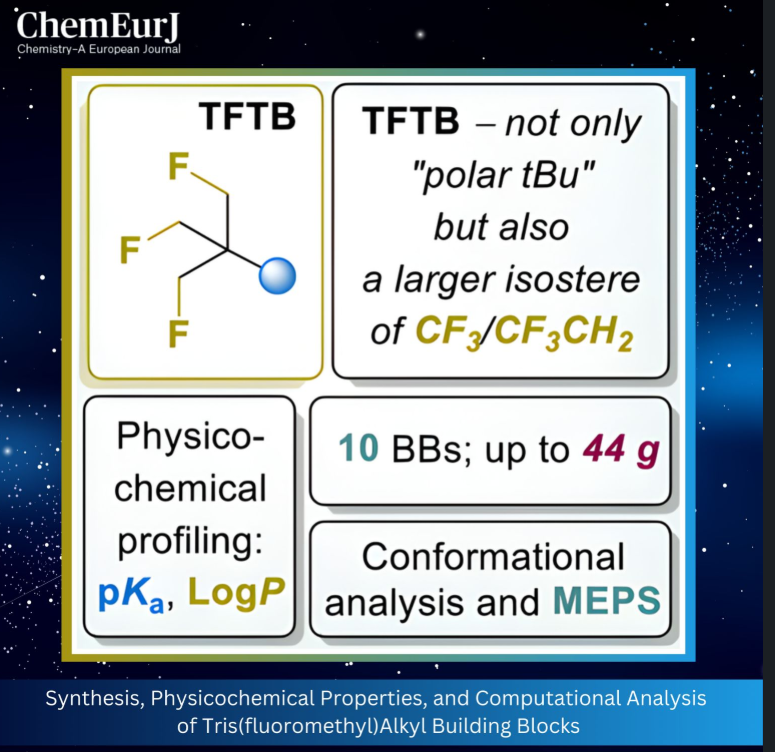
目前,氟原子、三氟甲基[7]、二氟亚甲基[8,9]、氟甲基[10]等经典含氟片段已得到深入研究,其对不同结构母核化合物理化性质的影响也已被广泛报道[11]。近年来,研究热点逐渐转向非经典含氟基团,这类基团能提供独特的空间位阻和电子特征,同时保持可预测的药代动力学行为[12,13]。其中具有代表性的包括O’Hagan课题组开发的多邻位氟代烷烃[14]、作为三氟甲基和乙基手性杂化生物等排体的1,2-二氟亚甲基基序(BITE基团)[15]、杂化偕二氟氧杂环丁烷基(diFOx)[16]与偕二氟环烷基团[17],以及各类常见官能团的改良型等排体[18,19]。
在各类非经典含氟基团中,近期重新受到关注的三(氟甲基)甲基片段(即β,β',β''-三氟叔丁基,TFTB,结构为(FCH2)3C)因独特的结构特征备受瞩目(图1B)[20,21]。该基团虽含有三个理论上具有柔性的C−CH2−F结构单元,但分子内固有静电相互作用能深度稳定其中一种构象,使其成为结构确定的叔丁基类似物[20,21]。在O’Hagan课题组发表上述结构分析之前,TFTB片段已在喹诺酮类抗生素系列中得到初步验证[22]:尽管该片段导致抗生素的抗菌活性有所下降,但其溶解度和脂溶性参数得到显著改善,凸显了该片段在生物等排性研究中的潜在价值。
尽管TFTB基团展现出良好的应用前景,但现有合成方法无法便捷制备适用于药物研发的多功能TFTB砌块。此前,O’Hagan课题组通过活化季戊四醇衍生底物的亲核取代反应,开发了芳基取代TFTB [20]和芳基-OCH₂-TFTB [21]化合物的合成方法;而胺类化合物1a的合成则需经过11步反应[23],步骤繁琐。
本研究以商用3,3-双(溴甲基)氧杂环丁烷(2)为起始原料,开发了克级规模制备TFTB取代砌块的实用方法,所得产物包括胺、羧酸、醇、卤化物及其高级直链同系物(图1C)。实验测定了模型衍生物质子化形式的酸性和脂溶性,揭示了α-杂原子取代TFTB衍生物相较于低氟取代叔丁基衍生物的独特性质。同时,通过计算分析解释了该类化合物的反常理化行为,并明确了碳连接链长度对其性质的影响。
3、结果与讨论
本研究最初计划通过氧化苯衍生物5制备羧酸4a(方案1)[20],其中化合物5可按O’Hagan课题组报道的方法,以苯乙醛为起始原料经三步反应合成,总产率37%[20]。
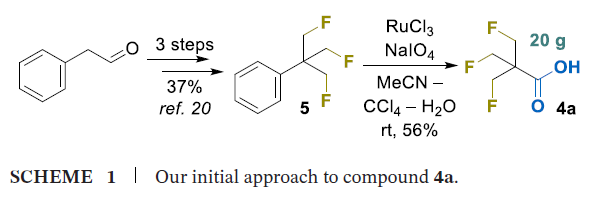
该方法虽能成功制备目标产物,但氧化步骤在放大生产时存在明显问题,因此本研究改用替代合成策略。新策略以氧杂环丁烷开环反应为基础,制备溴化物3a(方案2):将化合物2与氟化铯(CsF)在高温下反应,得到偕二氟甲基氧杂环丁烷6,分离产率69%;通过酸促进的氧杂环丁烷环开环反应,得到γ-溴代醇7,产率86%[24];最后采用四氟化硫/氟化氢(SF4/HF)混合体系对新戊醇5进行脱羟基氟化,得到目标产物 TFTB-CH₂Br(3a),分离产率68%。该合成路线可实现放大生产,单次反应可制备200g以上的化合物3a,且产率无显著下降。
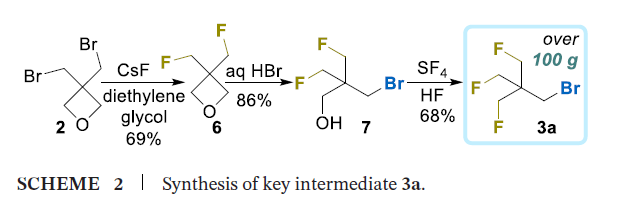
在获得大量溴化物3a后,本研究进一步制备了胺类同系物1a~c和羧酸类同系物4a~c(方案3):将溴化物3a与苯甲醇-氢化钠(BnOH-NaH)反应,得到醚类化合物8,在80g反应规模下产率73%;通过钯/碳(Pd/C)催化氢解反应得到醇9a,经升华纯化后产率85%;采用三氧化铬(CrO3)氧化醇9a,首次制得目标羧酸4a,单次13g反应规模下产率84%;通过改良的Curtius重排反应(以叠氮磷酸二苯酯DPPA为叠氮源)将羧酸转化为相应的N-苄氧羰基保护胺10,再经酸性脱保护,得到盐酸盐形式的胺1a,两步总产率72%,单次反应可制备20g。
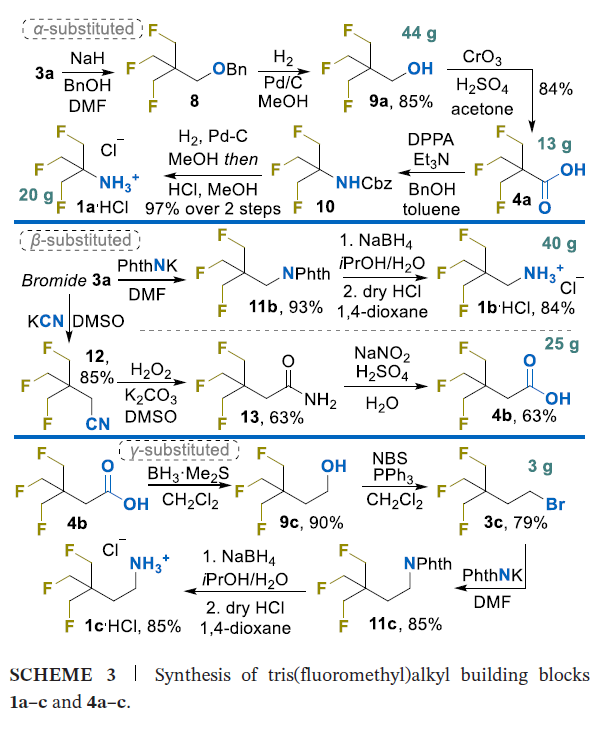
采用简便的化学反应,由溴化物3a制备得到同系物1b・HCl和4b:化合物3a与邻苯二甲酰亚胺钾反应,顺利得到衍生物11b;经硼氢化钠(NaBH4)还原,再用干燥氯化氢沉淀产物,得到盐酸盐1b・HCl,两步总产率78%,单次反应可制备40g。羧酸4b的合成以溴化物3a与氰化钾(KCN)在二甲基亚砜(DMSO)中100℃下的反应为起始,得到腈类化合物12,产率85%;尝试用盐酸水溶液、硫酸水溶液或碱直接水解腈基制备羧酸,均未获得成功;而采用过氧化氢(H2O2)在碳酸钾(K2CO3)存在下处理化合物12[25],可顺利得到酰胺13,产率良好;最后酰胺13在酸性条件下与亚硝酸钠(NaNO2)反应,得到目标羧酸4b,25g反应规模下产率63%。
以化合物4b为原料,经系列反应制备得到高级同系物1c・HCl和4c:采用硼烷-二甲硫醚络合物(BH3⋅Me2S)还原化合物4b,得到醇9c,产率90%;醇9c与N-溴代丁二酰亚胺-三苯基膦(NBS-PPh₃)反应,得到溴化物3c,产率79%;溴化物3c与邻苯二甲酰亚胺钾反应得到中间体11c(产率 85%),再经还原脱保护,得到盐酸盐形式的胺1c,产率85%。
3.1 酸性
本研究采用标准酸碱滴定法[16],测定了水溶液中21℃下TFTB取代的质子化胺1a~c、羧酸4a~c,单氟取代叔丁基(MFTB)衍生物15a、15b、19a,二氟取代叔丁基(DFTB)衍生物16a、16b、20a,以及无氟取代叔丁基(t-Bu)衍生物14a~c、18a、18b的酸解离常数(pKa)(图2);同时从文献中检索得到直链三氟甲基取代化合物17a~c、21a、21b的pKa数据[5,26,27]。
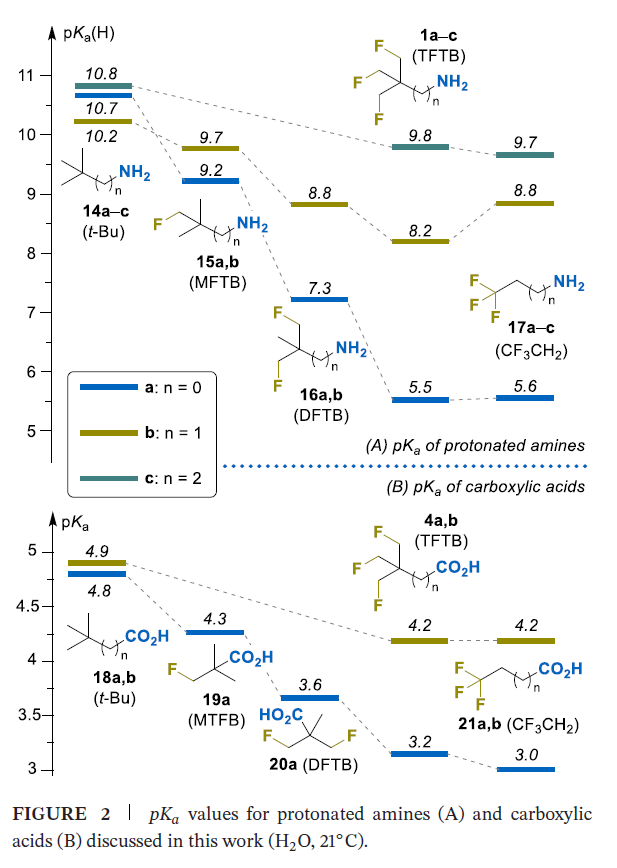
实验结果表明,含氟基团对化合物酸碱性质的净效应,与其与相应官能团之间的键连距离及氟原子数目密切相关。在叔丁胺(14a→15a→16a→1a)和新戊酸(18a→19a→20a→4a)衍生物系列中,叔丁基的逐步氟代使化合物的pKa值分别平均降低约1.7和0.5个单位/每个氟原子。当含氟基团与官能团之间引入亚甲基(CH2)后,氟原子的诱导效应显著减弱:在14b→15b→16b→1b系列中,每个氟原子导致的pKa降低值为0.8;而在14c→1c系列中,该数值仅为0.3。正如预期,在各类氟代叔丁基衍生物中,含氟原子数最多的TFTB衍生物,其质子化胺和羧酸的pKa值均为最低。从这一角度来看,TFTB基团与氟原子和官能团键连距离相同的2,2,2-三氟乙基(CF3CH2)取代基性质高度相似(对比1/17和4/21系列的pKa值)。
3.2 脂溶性
本研究采用经典摇瓶法结合定量高效液相色谱(HPLC)分析[16],测定了21℃下正辛醇-水体系中苯甲酰胺25a~c和苯胺26a、26b的脂水分配系数(Log P);其中苯甲酰胺25a~c由胺盐酸盐1・HCl与苯甲酰氯在三乙胺(Et3N)存在下反应制得,苯胺26a、26b由羧酸4与苯胺在1-乙基-3-(3-二甲氨基丙基)碳二亚胺(EDC)、4-二甲氨基吡啶(DMAP)和三乙胺存在下反应制得(图3)。
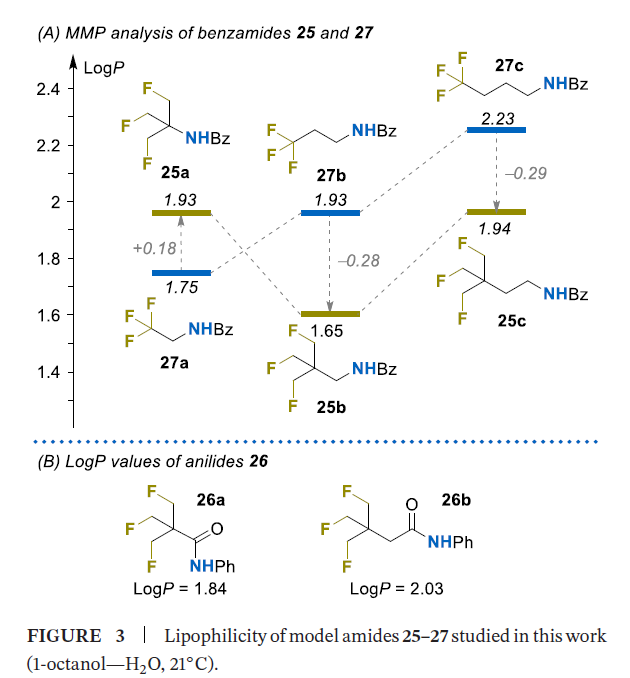
有趣的是,同系物苯甲酰胺25a~c的Log P值呈现非单调变化:25a(1.93)→25b(1.65)→25c(1.94)。该现象在三氟乙基取代的类似物27a~c及其他氟代衍生物中均未观察到[5],与无氟取代的直链N-烷基苯甲酰胺的变化规律也截然相反——后者每增加一个亚甲基,Log P值会规律性升高0.3~0.4个单位[5]。对上述所有同系物进行匹配分子对(MMP)分析发现,25a是该系列中的唯一特例,其脂溶性比三氟乙基取代的类似物27a高0.18个Log P单位;而25b、25c的脂溶性则比对应的27b、27c低0.28~0.29个Log P单位,这表明对于这两种化合物,TFTB取代基的脂溶性效应与三氟甲基基团更相似。值得注意的是,同分异构的苯胺26a、26b未出现上述反常规律,26b的Log P值比26a高0.19个单位。
3.3 构象分析
为解释化合物25a的反常行为,本研究对其进行了详细的构象分析:采用Grimme’s迭代流程结合静态元动力学模拟[28],以半经验紧束缚GFN2-xTB方法[29](CREST 软件实现[30])探索其构象空间;以最稳定构象为基础,研究单个C−CH2−F键全旋转坐标下的能量极值点(图4A,表S1),并在M062X/def2-TZVP水平[31]对各驻点进行结构优化;在相同理论水平下,通过频率计算得到标准温度和压力下的热力学校正值,结合非优化的旋转能量曲线,得到自由能变化值(ΔG或ΔG‡)(图4A,表S3)。
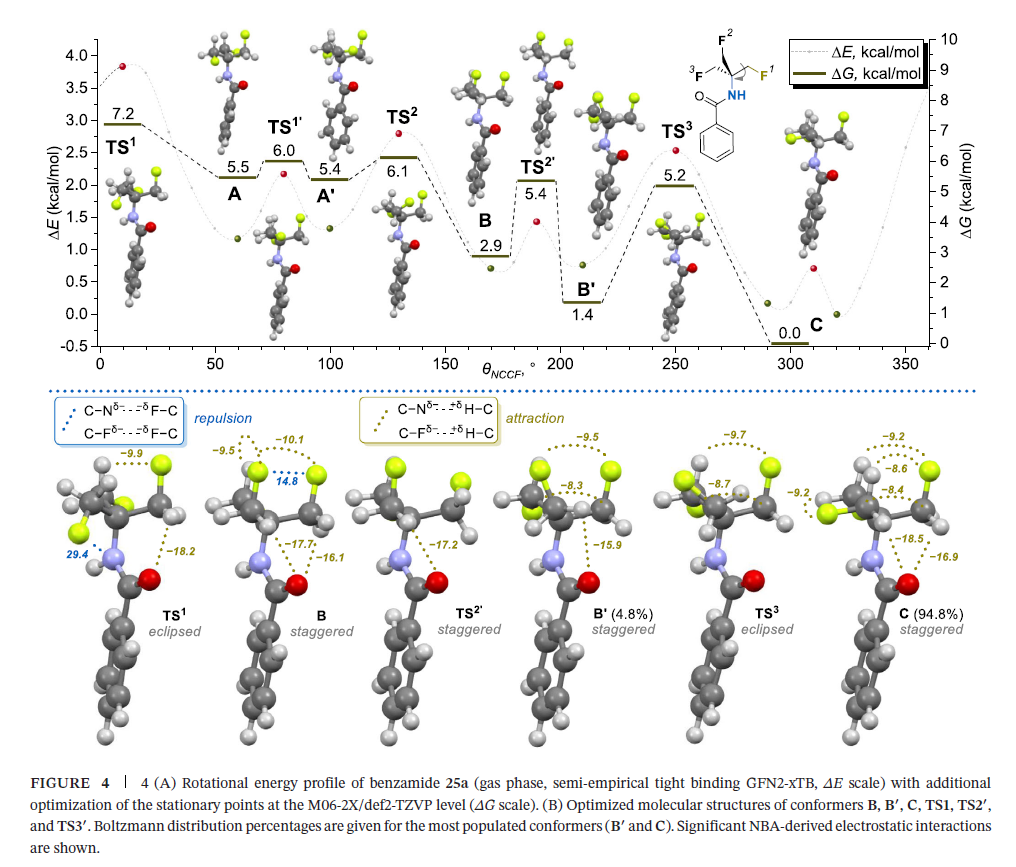
研究鉴定出三个对应重叠构象的旋转能垒(TS1−3),以及两个对应NCCF交错构象的额外旋转能垒。一维松弛扫描显示,构象C附近可能存在另一个能量最大值(TS3′),但无约束过渡态优化和内禀反应坐标(IRC)分析表明,该特征并非整个势能面上的一级鞍点;两个扫描最小值均收敛为单一构象C,说明TS3′是投影假象,其对应的能垒可忽略不计。
最高旋转能垒对应的过渡态TS1,其相对全局最低能构象C的自由能垒;次低能构象B′的能量比构象C高1.4 。根据各构象的相对能量和玻尔兹曼分布计算,气相中94.8%的分子采取全局最低能构象C,4.8%为构象B′,其余构象(A、A'、B)的总占比仅为0.4%。
在最稳定的构象C中,其中一个C-F键(C−F2)与C-N键呈反式共平面取向(图4B),另外两个C-F键与C-N键呈顺式斜交取向,二面角约为60°。该构象中,TFTB基团的两个C-H键与酰胺片段的C=O偶极近乎平行,这种取向产生了显著的静电稳定作用,与O’Hagan课题组此前发现的C−Fδ−⋅⋅⋅H−Cδ+有利相互作用协同增效[20,21]。此外,羰基与其中一个C-F键(C−F2)的反式定位可能形成初步的四中心键[32],该现象与CH2F片段中碳原子的电正性相关[33]。
本研究采用自然布居分析(NPA,NBO 7.0程序实现[34])得到原子自然电荷,结合经典库仑方程,鉴定了构象B、B'、C及过渡态TS1、TS2、TS3中所有显著的静电相互作用(包括稳定和去稳定作用)(表S2)。结果证实,除了奥黑根课题组发现的四个C−Fδ−⋅⋅⋅H−Cδ+相互作用(每个作用的稳定化能为-8.4~-9.2 )外,还存在两个C=Oδ−⋅⋅⋅H−Cδ+相互作用,其稳定化能分别为-18.5和-16.9 ,对构象稳定起到重要作用。而次低能构象B′中,未观察到构象C中的两个C−Fδ−⋅⋅⋅H−Cδ+和一个C=Oδ−⋅⋅⋅H−Cδ+相互作用,仅通过一个C=Oδ−⋅⋅⋅H−Cδ+(-15.9 )和两个C−Fδ−⋅⋅⋅H−Cδ+(-8.3、-9.2 )静电相互作用实现稳定;构象B′与C之间的过渡态TS3仅存在两个C−Fδ−⋅⋅⋅H−Cδ+相互作用,稳定化能为-8.7和-9.7 。
值得注意的是,上述所有过渡态中均未观察到C−Fδ−⋅⋅⋅C−Fδ−偶极-偶极去稳定相互作用;该相互作用仅在能量第三低的构象B中存在,但被C=Oδ−⋅⋅⋅H−Cδ+稳定相互作用抵消。能量最高的构象TS1中,平行的C−Nδ−与C−Fδ−偶极产生强排斥作用(28.6),是该构象能量较高的主要原因。
NPA分析解释了旋转能量曲线中交错过渡态TS1′和TS2的存在:这些物种虽无上述偶极-偶极去稳定相互作用,但破坏了所有低能构象中均存在的C=Oδ−⋅⋅⋅H−Cδ+稳定相互作用,导致自由能升高;该现象也是构象B和B′能量相对较高的原因之一,其中构象B′的C=Oδ−⋅⋅⋅H−Cδ+相互作用抵消了C−Fδ−⋅⋅⋅C−Fδ−偶极-偶极排斥作用的影响。上述结果表明,酰胺片段中的杂原子(氧、氮)对TFTB优势构象的稳定起到关键作用,这也使α-杂原子取代TFTB衍生物成为药物研发中极具特色的分子片段。
3.4 分子静电势表面分析
从取代基的范德华体积来看,TFTB片段与叔丁基接近(分别为74.1和73.5)[35],远大于三氟甲基(42.8)和三氟乙基(60.1);因此,无法通过简单的体积因素解释TFTB衍生物的脂溶性特征[36]。为解决这一问题,本研究以25a的最稳定构象为基础,对同系物苯甲酰胺25a~c进行分子静电势表面(MEPS)分析。
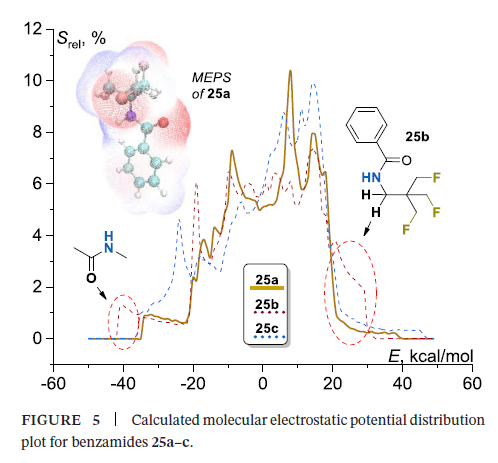
Hunter课题组此前的研究表明,气相中计算得到的分子静电势表面极值分布与相转移自由能相关,而相转移自由能又通过特定的溶质-溶剂相互作用与Log P值直接关联[37,38]。本研究借鉴该方法[39,40],计算了化合物25a~c的分子静电势表面,并通过绘制相对表面积-电势值曲线,分析了分子表面的电势分布(图5)。对比结果显示,各同系物的分子静电势分布存在明显差异:脂溶性最低的25b在低电势区(-40~-35)和高电势区(20~40)的表面积占比显著更高,而25a和25c在上述两个电势区的分布则较为相似。
该结果在一定程度上与实验测得的Log P变化规律相符:25a和25c的分子静电势分布相似,而25c含有两个额外的亚甲基,理论上应具有更高的脂溶性。进一步分析发现,25b的高电势区对应亚甲基上高度极化的C-H键,该效应在25a中完全不存在,在25c中也显著减弱。上述发现为25a脂溶性较高的现象提供了合理的解释:一方面,25a中不存在同时受酰胺和TFTB基团直接/间接极化的C-H键(如 25b中的此类键);另一方面,氟原子的强诱导效应降低了羰基的极化程度(低电势区的静电势分布可佐证),最终导致分子整体极性降低。此外,羰基氧原子的氢键形成能力[41]也因上述极化重排而下降,这也是其脂溶性升高的另一原因。
4、结论
本研究以商用氧杂环丁烷衍生物为起始原料,建立了三氟甲基烷基(即β,β',β''-三氟叔丁基,TFTB)取代砌块的简洁、可规模化合成路线,制备得到含氨基(NH2)、羧基(COOH)、羟甲基(CH2OH)、溴甲基(CH2Br)等官能团的衍生物。本研究的关键中间体(FCH2)3CCH2Br经三步反应合成,总产率40%,且可放大至200g规模,产率无显著下降。
通过实验测定离子化基团的酸性(pKa)和模型苯甲酰胺衍生物的脂溶性(Log P)等理化参数发现,TFTB取代基本非叔丁基的简单等体积替代物,而是类三氟甲基/三氟乙基的极性调节剂,其对pKa和Log P的调节效应介于两者之间。构象研究表明,TFTB取代基倾向于采取螺旋状空间构型;除了已有文献报道的C−F⋅⋅⋅H−C相互作用对构象平衡的影响外,C=O⋅⋅⋅H−C静电相互作用对该优势构象的稳定也起到重要作用。
总体而言,杂原子取代TFTB基序兼具强局部诱导效应、可预测的构象偏好,以及类三氟甲基/三氟乙基的酸碱性-脂溶性特征,同时其空间位阻体积高达后两者的两倍,这使该取代基成为药物设计中极具前景的生物等排取代片段。
本文中涉及的含三(氟甲基)烷基砌块与相关衍生物,Enamine均有现货供应,如需了解更多可访问官网查询:https://www.enamine-genez.com/