1、摘要
先说结论:真实存在
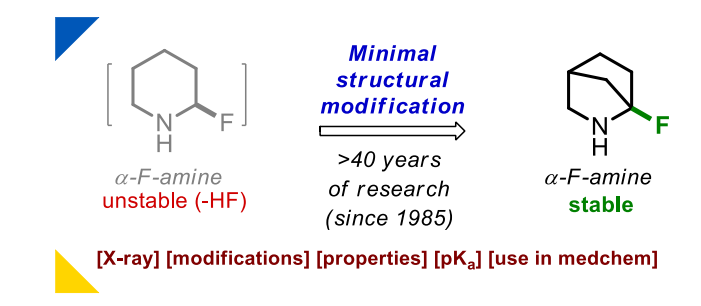
1985年以来,科学家们一直致力于脂肪族α-氟胺的合成研究,然而这类结构因易发生脱氟化氢反应而极不稳定。本研究证实,对难以合成的α-氟哌啶进行最小化结构修饰——引入亚甲基(CH₂)基团,可得到稳定的α-氟胺。这一此前未被发现的化合物类别已得到全面的研究与表征,并成功应用于一项药物化学研究中。
2、引言
哌啶是药物分子中最常见的三大环系之一。1975年,默克公司的科洛尼奇及其团队首次合成了γ-氟哌啶,此后β-氟哌啶和γ-氟哌啶在化学领域中占据了重要地位,其结构母核出现在超10000项各大制药企业的专利中。从分子层面来看,向哌啶环中引入氟原子可稳定其构象,并降低氮原子的碱性。
氟原子引入哌啶环可得到三种同分异构体:α-、β-和γ-氟哌啶。与性质稳定的β-和γ-异构体截然不同,α-氟哌啶的合成始终未获成功。1985年起,科学家们多次尝试制备N-未保护的脂肪族α-氟胺,但这类化合物均因易发生脱氟化氢反应而不稳定。2003年,科特拉团队终于合成了该类化合物的首个代表物——苄基氨基氟乙酸乙酯,不过研究人员指出,该化合物“稳定性极差,无法参与任何化学反应”。
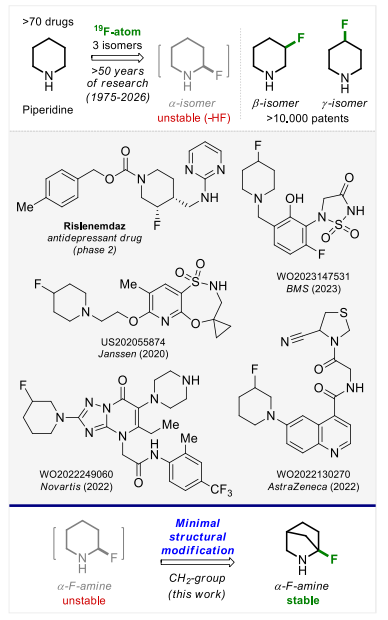
本研究证实,对难以合成的α-氟哌啶进行最小化结构修饰(引入CH₂基团),可获得稳定的α-氟胺。这一此前被忽视的化合物类别已完成全面表征与化学修饰,并成功应用于药物化学研究。该发现有望对药物研发领域产生积极影响,为药物化学家开发新型药物提供新的思路。
3、结果与讨论
意外发现
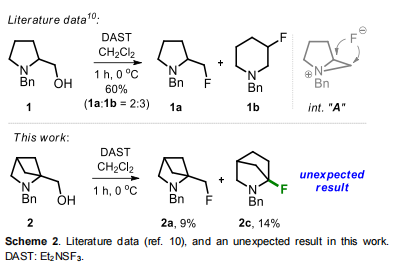
已有研究表明,脯氨醇1与二乙胺基三氟化硫(DAST)反应会生成异构体产物1a和1b的混合物,该反应通过阳离子氮杂环丙烷鎓中间体A进行。本课题组此前的研究证实,2-氮杂双环[2.1.1]己烷是药物化学中一类实用的合成砌块。本研究中,氨基醇2与DAST反应时,意外生成了N-取代α-氟哌啶2c(产率14%),同时还得到了异构体2a(产率9%)。
合成工艺优化
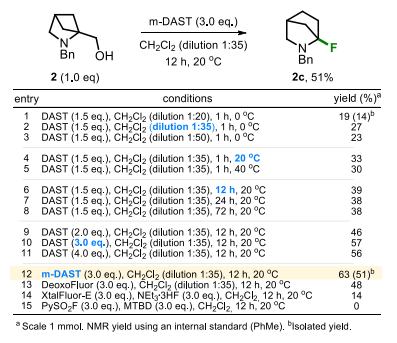
2c的生成让研究团队备受启发,因此进一步对该产物的合成产率进行优化。通过调整反应稀释比、温度、反应时间及DAST的用量,产物的核磁共振产率从19%提升至57%;对不同氟化试剂进行筛选后发现,吗啉基三氟化硫(mDAST)是最适宜的氟化试剂。在优化后的反应条件下,化合物2c的分离产率最终达到51%。
底物适用范围
在2c的合成工艺优化完成后,研究团队进一步探究了取代2-氮杂双环[2.1.1]己烷12在该反应中的反应性。研究发现,带有给电子取代基的底物3-7能以57%~73%的较高产率生成N-取代α-氟哌啶3c-7c;而带有吸电子取代基的底物8-10主要生成常规的脱氟羟基化产物8a-10a,仅能检测到微量的α-氟胺衍生物c。值得注意的是,所有实验中均未检测到3-氟哌啶异构体b的生成。
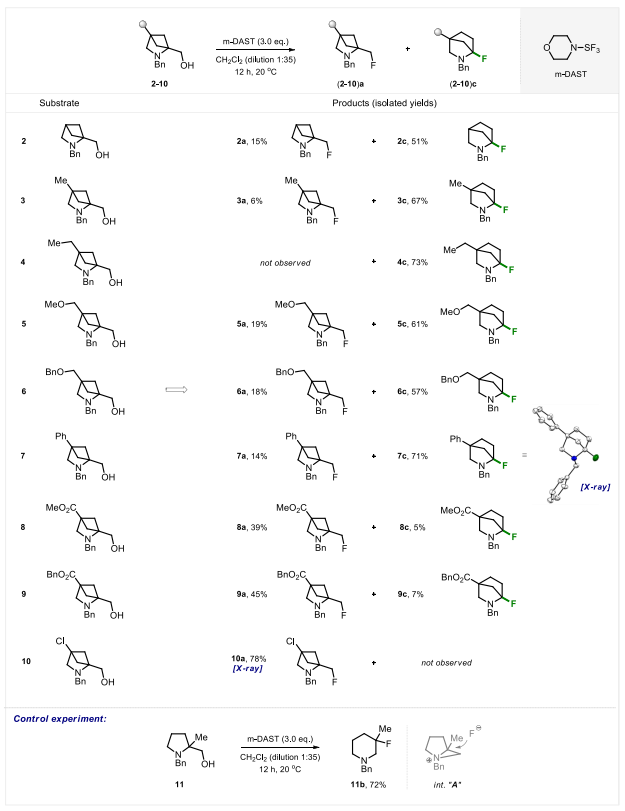
以醇11为底物进行对照实验,仅得到了与文献报道一致的β-氟哌啶11b。上述实验结果表明,2-氮杂双环[2.1.1]己烷母核和给电子取代基的存在,是N-取代α-氟胺c生成的关键条件。
反应机理
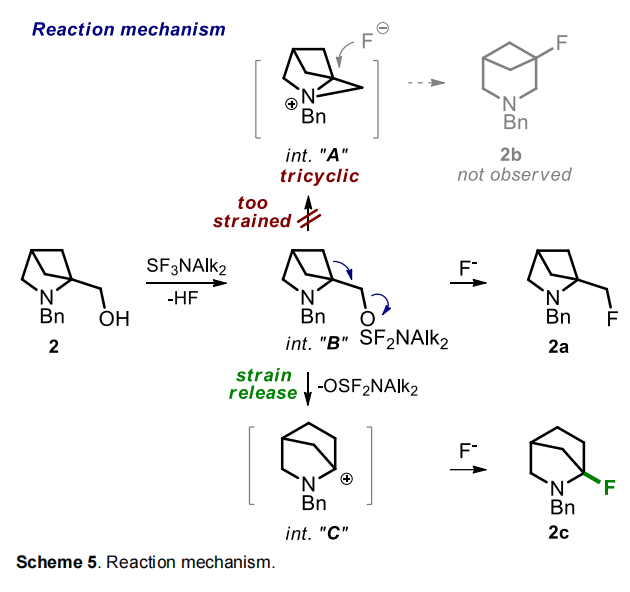
本研究提出的反应机理可合理解释上述实验结果:氨基醇2与mDAST反应生成常规中间体B,氟离子对中间体B中的离去基团进行亲核取代(SN2),生成产物2a。与单环醇1不同,双环醇2不会形成具有张力的三环中间体A(与文献报道一致),这也是底物2-10均未生成3-氟哌啶b的原因。随后,反应发生“张力释放型”扩环反应,生成中间体C——此前研究认为该中间体无法形成。氟离子与中间体C进一步反应,最终生成α-氟哌啶c。给电子取代基可促进该扩环步骤,而吸电子取代基则会抑制该反应,与实验结果相符。
N-未保护 α-氟胺的合成
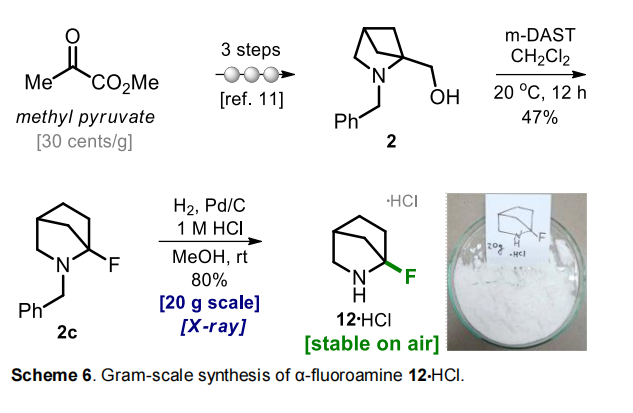
尽管已知N-未保护的脂肪族α-氟胺稳定性极差,本研究团队仍出于纯科学探索的目的,尝试对化合物2c进行N-脱保护反应。令人意外的是,在甲醇的酸性体系中,采用氢气/钯炭对2c进行常规氢化反应,得到了一种白色结晶固体。X射线晶体衍射分析证实,该产物为盐酸盐形式的N-未保护α-氟哌啶12。
产物12・HCl在空气中表现出出人意料的稳定性,因此研究团队进一步实现了该化合物的克级合成。以廉价易得的商用丙酮酸甲酯为原料,经三步反应制得醇类前体;该前体与mDAST在上述优化条件下进行克级反应,以47%的产率得到产物2c;2c在酸性条件下脱苄基,以80%的产率得到目标胺12・HCl。采用该工艺,单次反应可轻松制备20克产物。
α- 氟胺的化学修饰
本研究对-氟哌啶12・HCl的化学性质进行了系统探究,发现其能在常规条件下顺利参与所有胺类的典型反应:磺酰胺合成(产物13)、酰胺合成(产物14)、烷基化(产物15)、氨基甲酸酯合成(产物16)、脲合成(产物17)、Chan-Lam型芳基化(产物18)以及芳香族亲核取代(SNAr)芳基化(产物19),其中产物17和19的结构经X射线晶体衍射确证。
研究团队还尝试制备胺12的碳位取代衍生物:化合物6c在酸性条件下经氢气/钯炭氢化,实现N-苄基和O-苄基的同时脱除,得到另一种稳定的α-氟哌啶20・HCl;20・HCl经叔丁氧羰基(Boc)保护得到氨基醇21;21的羟基经斯文氧化生成醛22,醛22再经大平-贝斯特曼炔基化反应得到氨基炔23;23经常规酸性脱Boc保护,生成α-氟哌啶24・HCl;21的羟基经高碘酸钠/三氯化钌氧化,得到N-Boc保护的氨基酸25;25经酸性脱Boc保护,得到另一种稳定的α-氟胺26・HCl。
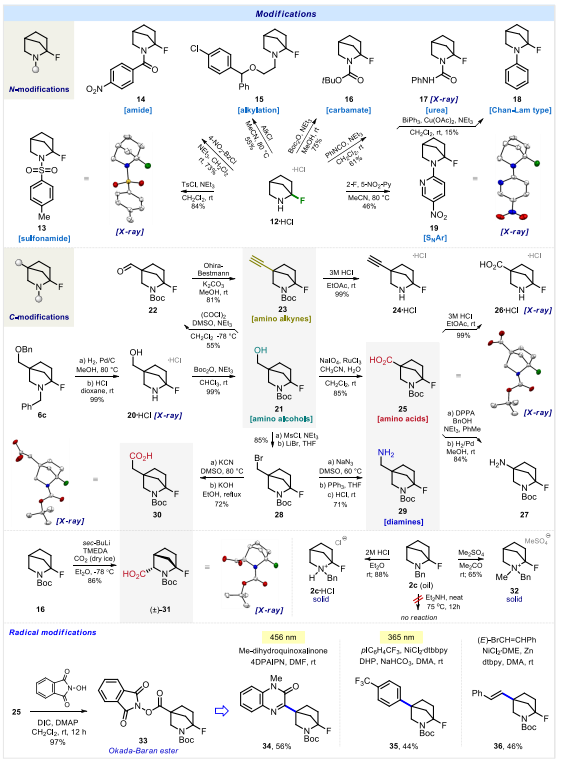
羧酸25经Curtius重排反应后,其生成的中间体再经N-苄氧羰基(Cbz)氢化脱除,得到N-Boc保护的二胺27;21的羟基经甲磺酰化后,与溴化锂发生亲核取代,得到溴代物28;28与氰化钾加热反应,再经碱水解,得到N-Boc保护的氨基酸30。化合物16与仲丁基锂/四甲基乙二胺反应后,通入干冰,单一立体异构体的N-Boc保护氨基酸31得以生成。其中,化合物20・HCl、25、26・HCl和31的结构均经X射线晶体衍射确证。
值得注意的是,尽管化合物2c的α-位存在吸电子的氟原子,其氮原子仍能轻松发生质子化甚至季铵化反应,生成的产物2・HCl和32均为稳定的结晶固体。
本研究还实现了α-氟哌啶骨架的自由基修饰:羧酸25与N-羟基邻苯二甲酰亚胺在N,N'-二异丙基碳二亚胺/4-二甲氨基吡啶的作用下,于二氯甲烷中室温反应,生成Okada-Baran中间体33;33与1-甲基二氢喹喔啉酮发生Minisci反应,以56%的产率得到化合物34;在Hantzsch存在下,33与对三氟甲基碘苯经光催化镍催化的sp³-碳-sp²-碳交叉偶联反应,生成产物35;33与反式-β-溴苯乙烯在锌粉存在下经光催化镍催化偶联,生成烯烃36。
产物的稳定性
与文献报道中不稳定的N-未保护脂肪族α-氟胺截然不同,本研究合成的所有化合物均具有良好的稳定性,其中大部分为白色结晶固体,可在室温空气中储存。
α-氟哌啶母核甚至能耐受仲丁基锂等苛刻的反应条件;N-未保护的α-氟哌啶12・HCl和20・HCl与各类碱性体系完全相容,无任何脱氟化氢的迹象。此外,针对胺2c中氟原子的取代尝试均未成功,例如将2c在过量二乙胺中75℃加热12小时,未检测到任何反应,仅能回收起始原料。
所有产物在密封样品瓶中室温储存,经三个月的定期核磁共振和液质联用检测,均未发现可检测到的分解现象。
本研究合成的N-未保护α-氟哌啶具有高稳定性,其原因可通过布雷特规则解释:小双环体系的桥头位置无法形成双键,因此该类化合物无法发生脱氟化氢反应(该反应需形成双键)。
α-氟胺在药物分子中的引入及理化性质
鉴于α-氟胺出人意料的高稳定性,研究团队进一步探究了α-氟化修饰对哌啶类化合物理化性质的影响。为此,本研究合成了三种模型化合物和两种药物的α-氟化衍生物,并引入2-氮杂双环[2.2.1]庚烷衍生物作为对照(该结构为药物化学中已成熟应用的母核)。所选的生物活性分子为抗组胺药氯哌斯汀和天然产物胡椒碱,研究对象分为N-烷基化哌啶和N-酰基化哌啶两类。
水溶性
在N-烷基化化合物37和氯哌斯汀中,将哌啶母核替换为α-氟胺后,水溶性显著下降约50%:37的水溶性为391微摩尔/升,而2c为235微摩尔/升;氯哌斯汀的水溶性为327微摩尔/升,而其衍生物15为200微摩尔/升。
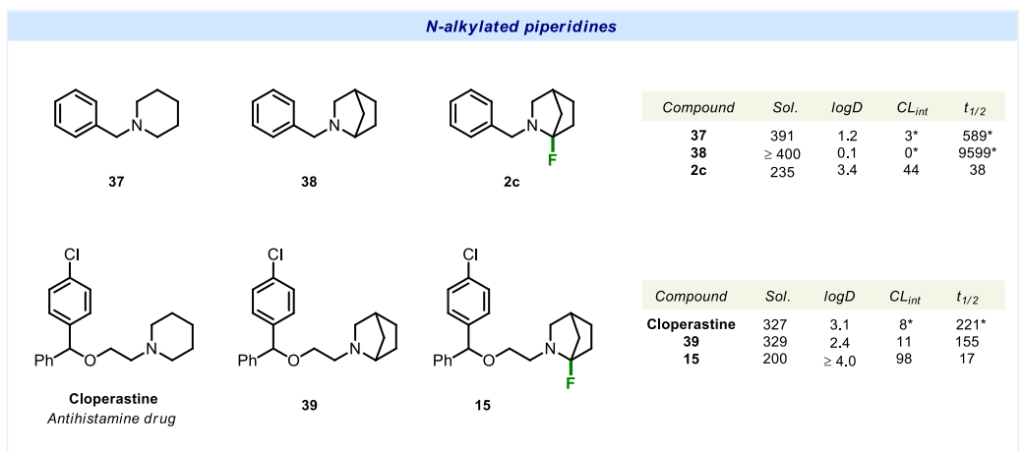
在N-酰基化化合物40、43和胡椒碱中,该结构替换对水溶性的影响无明显规律:酰胺40的水溶性过高,超出实验方法的检测范围,因此未检测到明显变化;酰胺43的水溶性下降至原来的1/3(43为369微摩尔/升,45为116微摩尔/升);而胡椒碱经修饰后,水溶性大幅提升6倍(胡椒碱为40微摩尔/升,衍生物47为244微摩尔/升)。
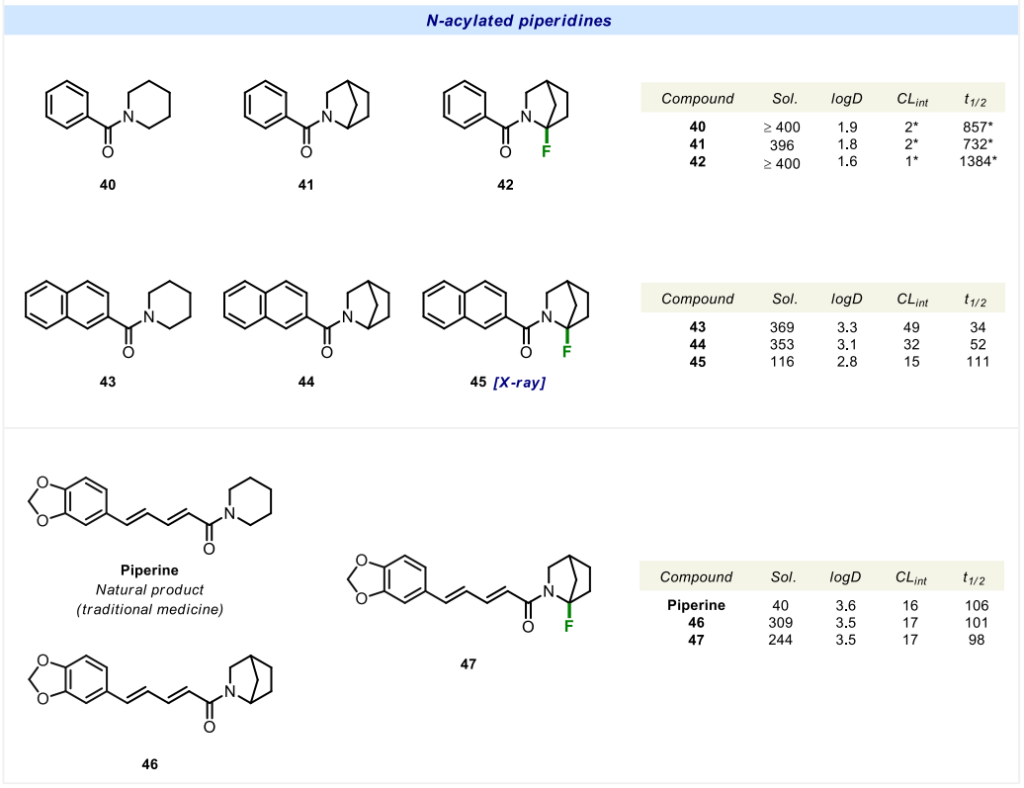
脂溶性(logD)
在所有五种研究对象中,将哌啶母核替换为2-氮杂双环[2.2.1]庚烷后,脂溶性均有所降低:N-烷基化哌啶37和氯哌斯汀的logD值下降约0.7~1.1个单位,N-酰基化哌啶40、43和胡椒碱的logD值下降幅度较小,仅0.1~0.2个单位。
在2-氮杂双环[2.2.1]庚烷母核基础上进一步引入α-氟原子,对N-烷基化和N-酰基化哌啶的脂溶性产生了截然不同的影响:N-烷基化化合物的脂溶性大幅提升,logD值远超未取代的原哌啶化合物(37:1.2;38:0.1;2c:3.4;氯哌斯汀:2.1;39:2.4;15:≥4.0);而在两种N-酰基化化合物中,α-氟原子的引入使脂溶性进一步降低(40:1.9;41:1.8;42:1.6;43:3.3;44:3.1;45:2.8);第三种N-酰基化化合物胡椒碱的脂溶性则无明显变化(胡椒碱:3.6;46:3.5;47:3.5)。
代谢稳定性
在所有五种研究对象中,将哌啶母核替换为2-氮杂双环[2.2.1]庚烷后,代谢稳定性无明显变化。与脂溶性的变化规律相似,进一步引入α-氟原子对N-烷基化和N-酰基化哌啶的代谢稳定性影响迥异:N-烷基化哌啶的代谢稳定性显著下降,成为代谢易降解型化合物(2c的肝微粒体清除率CLint=44微升/(分钟・毫克);15的CLint=97微升/(分钟・毫克));N-酰基化哌啶40因本身稳定性极高,α-氟原子的引入未对其代谢稳定性产生可检测的影响;N-酰基化哌啶43经α-氟化修饰后,代谢稳定性显著提升(43的CLint=49微升/(分钟・毫克);44的CLint=32微升/(分钟・毫克);45的CLint=15微升/(分钟・毫克));而胡椒碱经α- 氟化修饰后,代谢稳定性无明显变化(胡椒碱的CLint=16微升/(分钟・毫克);46的CLint=17微升/(分钟・毫克);47的CLint=17微升/(分钟・毫克))。
小结:将哌啶母核替换为α-氟胺后,N-烷基化化合物的药物化学相关理化性质发生负面变化:水溶性下降、脂溶性升高、代谢稳定性降低;而N-酰基化化合物的水溶性无明显变化,脂溶性呈有益降低趋势,且代谢稳定性保持良好。
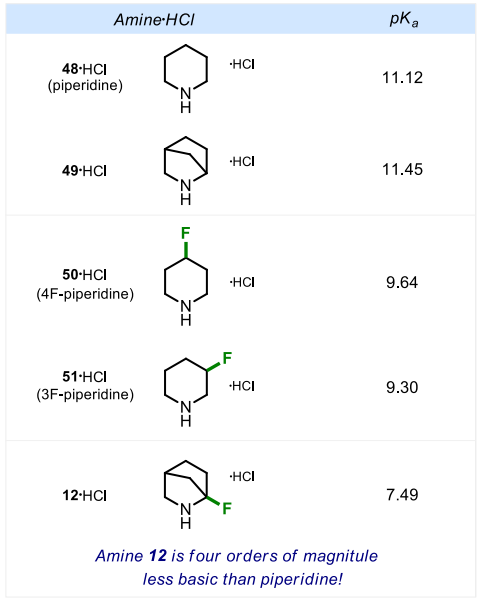
酸解离常数(pKa)
生物活性分子中的碱性氮原子是导致HERG通道毒性的常见原因,在氮原子附近引入吸电子的氟原子是降低其碱性的常用策略,该修饰仅会对分子进行最小化的结构改变,这也是3-氟哌啶和4-氟哌啶在现代药物研发中应用广泛的原因。
哌啶(48)中引入亚甲基得到化合物49后,氮原子的碱性略有升高;而哌啶的β-位和γ-位引入氟原子得到50和51后,氮原子的碱性降低两个数量级;在2-氮杂双环[2.2.1]庚烷的α-位引入氟原子得到12后,氮原子的碱性降低四个数量级。值得注意的是,尽管碱性大幅降低,胺12及其衍生物仍能在常规条件下完成氮原子的各类修饰反应。
生物活性
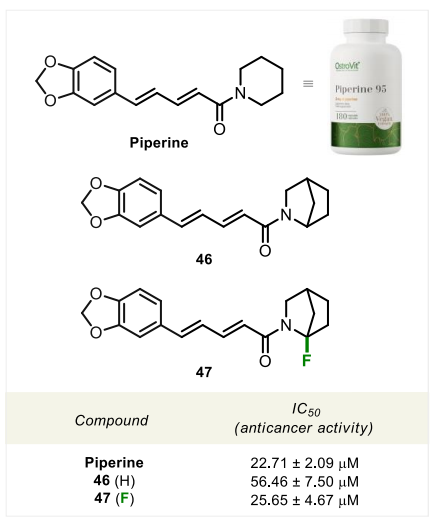
近期研究证实,胡椒碱在乳腺癌治疗中展现出良好潜力,能显著抑制4T1乳腺癌细胞的增殖,并有效抑制肺癌转移。然而,胡椒碱的水溶性极差,这一缺陷使其难以进入临床应用。
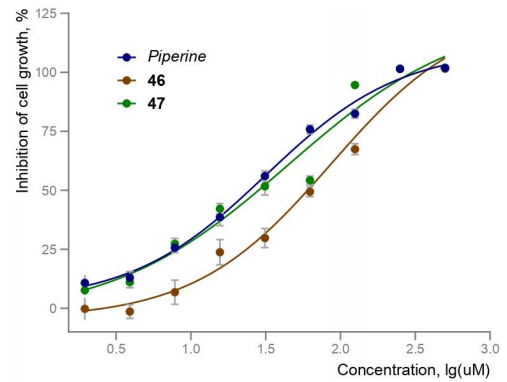
本研究中,胡椒碱的α-氟化衍生物47的脂溶性和代谢稳定性与原分子基本一致,而水溶性大幅提升600%。基于此,研究团队进一步探究了衍生物47的生物活性,采用刃天青还原法检测细胞呼吸抑制情况,研究了胡椒碱及其类似物46和47对小鼠4T1乳腺癌细胞的细胞抑制作用。
刃天青还原实验结果显示,胡椒碱对4T1细胞表现出中等抑制活性(半数抑制浓度IC₅₀=22.71±2.09微摩尔/升);其双环类似物46的活性降低约一半(IC₅₀=56.46±7.50微摩尔/升);而氟化衍生物47的活性与胡椒碱几乎一致(IC₅₀=25.65±4.67微摩尔/升),且解决了胡椒碱水溶性差的问题。
上述结果证实,性质稳定的N-酰基化α-氟哌啶类化合物完全适用于药物研发项目。
4、结论
哌啶是药物研发中最核心的三大环系之一,氟原子引入哌啶环可得到α-、β-、γ-三种异构体。其中β-和γ-氟哌啶已在化学领域中得到广泛应用,而α-氟哌啶的合成长期未获成功。1985年以来,科学家们一直尝试制备N-未保护的脂肪族α-氟胺,却始终受限于其不稳定性。
本研究证实,对难以合成的α-氟哌啶进行最小化结构修饰——引入亚甲基基团,可得到化学性质稳定的α-氟胺12,该化合物可在室温空气中稳定储存。胺12的碱性较哌啶降低四个数量级,同时仍保留足够的化学反应活性,能在常规条件下参与所有胺类的典型反应(烷基化、酰基化、磺酰胺合成、脲合成、芳香族亲核取代等)。
此外,α-氟胺12的N-烷基化衍生物代谢稳定性较差,而N-酰基化衍生物则具有良好的代谢稳定性;其中氟化衍生物47的抗乳腺癌活性与胡椒碱相当,且水溶性较天然产物胡椒碱提升600%。
本研究团队希望,这一此前被忽视、难以合成的α-氟胺类化合物,能在未来的药物研发项目中得到广泛应用,为药物化学家开发更优质的新型药物提供新的方向。