计算机辅助药物设计
以精准和洞察力加速药物发现
通过Enamine先进的计算机辅助药物设计(CADD)服务,释放体外药物发现的全部潜力。
与我们合作,您将获得量身定制的建模工作流程,整合了先进的分子建模、人工智能驱动的筛选以及药物化学专业知识——所有这些都专注于提高您在早期药物发现中的成功率。
我们的团队将全程支持您,从靶点识别和结构预测,到命中化合物识别、先导化合物优化,直至后续开发。我们结合了基于结构和基于配体的虚拟筛选、分子对接、分子动力学以及自由能微扰(FEP)等工具,帮助您优先选择最有前景的候选化合物进行合成和生物测试。
您获得的不仅仅是计算工具的使用权,更是一种以结合亲和力预测、ADMET特性分析和实时决策为核心的结果导向型策略,旨在降低您研发管线的成本和时间。
通过与Enamine CADD部门合作,您将受益于:
- 对靶点行为和结合口袋动态的深入洞察
- 从Enamine REAL库和筛选库中快速识别和排序命中化合物
- 整合人工智能(AI)、机器学习(ML)和化学信息学,探索广阔的化学空间
- 从虚拟筛选到合成及生物验证的无缝衔接
无论您面对的是具有挑战性的蛋白质、RNA结构,还是新颖的化学系列,我们的计算化学专业知识都能帮助您自信地进行设计、预测和优化。
我们采用平衡的方法,综合运用多种互补的技术和策略
靶点建模与研究
同源建模
- 针对未解析结构的靶点进行三维结构预测
- 结构修复
- 突变体建模
靶点结构分析
- 潜在结合位点搜索
- 活性残基预测
- 人工智能结合点位预测
DNA/RNA-复合物建模
- RNA 沟槽中的嵌插与结合
- G-四链体(G-quadruplex)建模
- 核糖开关(Riboswitches)
- DNA GG 和 GA 结合口袋
分子动力学(MD)模拟
- 比较并展示配体存在与不存在时结构随时间变化的行为
- 模拟突变对蛋白稳定性、活性以及对已知结合物抗性的影响
- 蛋白-配体结合能估算
- 隐蔽性结合口袋(Cryptic pocket)搜索
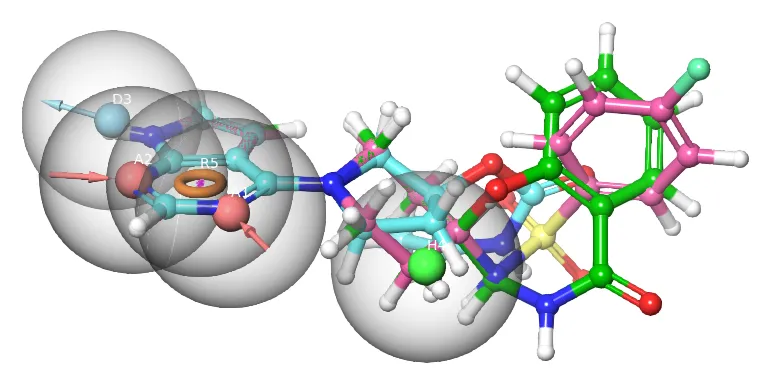
基于配体的药物设计(LBDD)
类似物搜索与子结构搜索
- 二维搜索(摩根指纹、ECFP等)
- 三维搜索(QuickShape)
- 多样化/聚类分析
- Bemis-Murcko骨架/图谱搜索
- 合成子搜索
基于配体的三维药效团虚拟筛选
机器学习建模与预测
- 机器学习驱动的高通量虚拟筛选
- 机器学习辅助构建QSAR模型
- 机器学习驱动的ADMET性质预测
命中化合物到先导化合物的集成项目支持
- 合成可行性评估与筛选结果分析
- 构效关系(SAR)
- 骨架跃迁/骨架变换
- 生物电子等排体替换
电子结构计算:DFT、MP2等
QM/MM模拟
大规模数据可视化
- UMAP
- PMI-3D
- 性质分布图
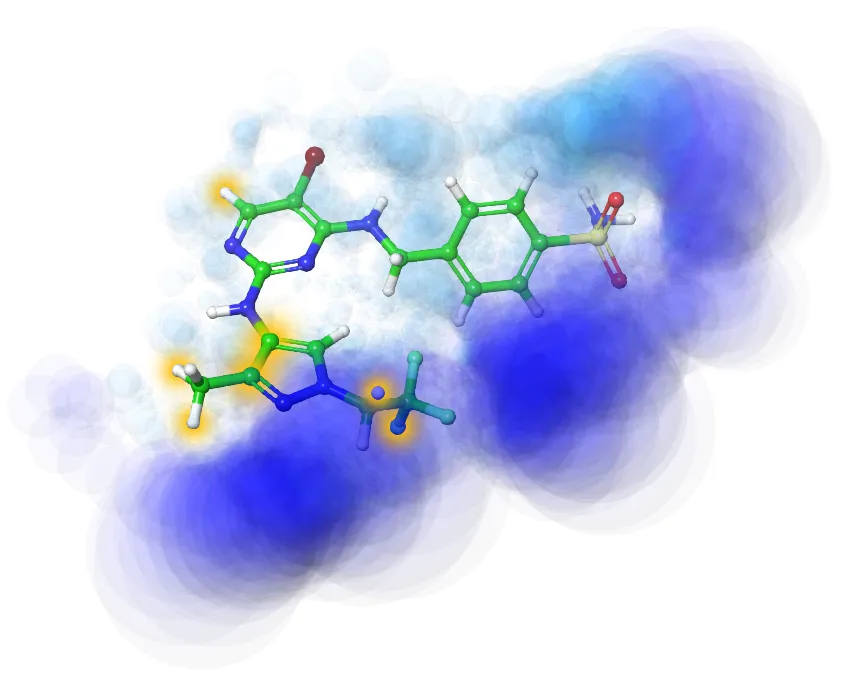
性质计算
- 毒性/ADMET机器学习预测(BBB、hERG、渗透性(Caco-2、PAMPA等)、代谢稳定性预测等)
- 理化/药物化学性质预测与分析(logP/logD、溶解度等)
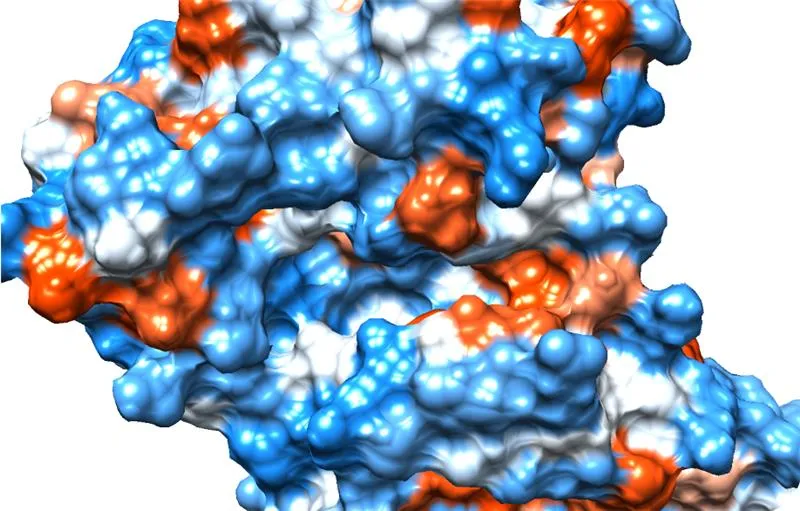
基于结构的虚拟筛选(SBDD)与复合物评估
非共价分子对接
- 经诱饵验证的模型
- 定制化约束条件
- 多阶段工作流程
共价分子对接
- 预设化学反应及靶向设计化合物库
- 多阶段工作流程(+非共价对接用于反应前建模)
基于片段的虚拟筛选
基于主动学习的建模
Boltz-2 方法
- 结构预测
- 结合模式确定
- 结合概率估计
- 结合亲和力预测
汤普森采样(Thompson Sampling)
- 自适应化合物筛选策略
- 指导化学空间的探索
- 高效优化排序多样化且有前景的候选分子
元动力学(Metadynamics)
- 用于稀有事件的增强采样技术
- 高效识别自由能极小值和过渡态
- 探索隐藏的构象空间
- 支持结合构象优化与作用机制解析
分子动力学(MD)模拟
- 验证虚拟筛选中选出的配体
- 估算蛋白-配体结合能
- 提供蛋白-配体相互作用的动态图像
QM/MM 计算
- 量子力学/分子力学混合建模
- 精确描述反应机制
- 精细化模拟共价键形成与电荷转移过程
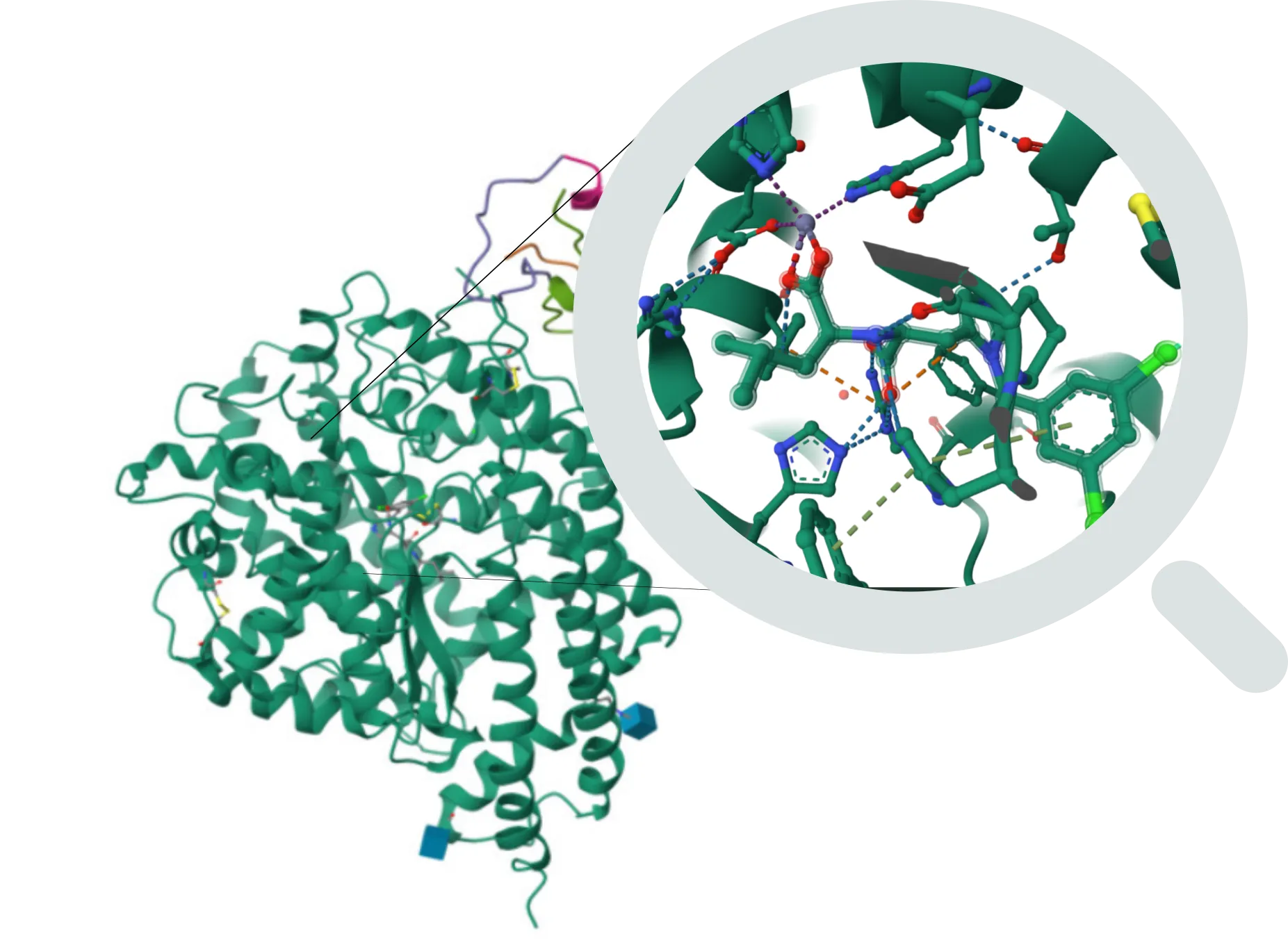
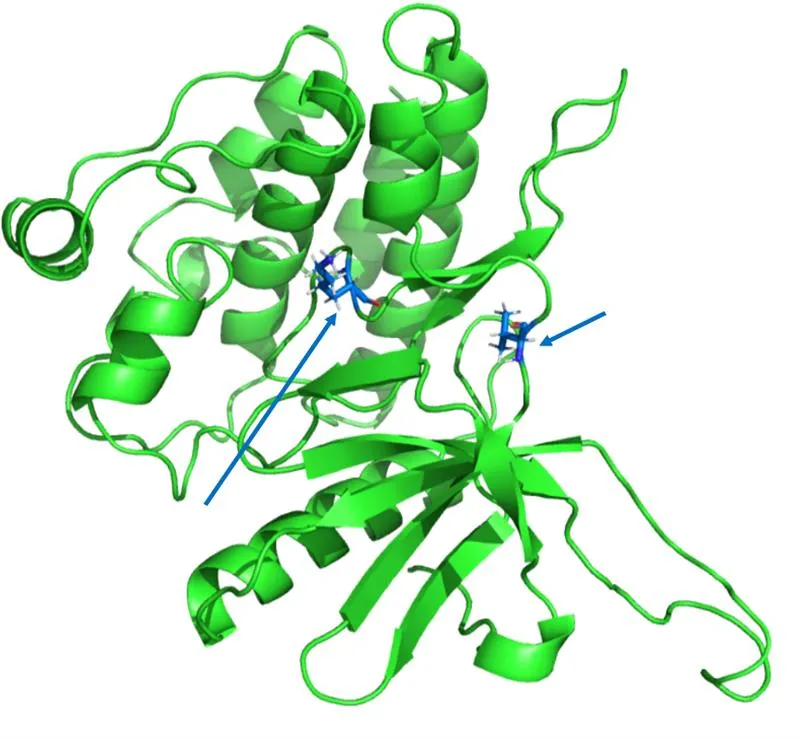
新方法
FEP(自由能微扰)- 精确结合亲和力预测
它解决的问题:
- 精确计算配体与靶标的结合亲和力
- 通过结合自由能对化合物进行排序,以改进构效关系(SAR)分析
- 预测修饰对结合亲和力的影响
- 通过选择最有潜力的候选化合物,降低合成和生物测试的成本

提升配体设计的全新准确水平!
每个项目都始于评估阶段
在启动全面的建模工作之前,我们始终会先进行项目评估阶段——这是一个简明的分析步骤,旨在:
- 为虚拟筛选或先导化合物优化制定科学合理的策略
- 评估可用结构数据(如靶点、配体等)的充分性与适用性
- 根据您的目标和限制,推荐最有效的方法和工作范围
- 识别早期药物发现项目中潜在的风险或挑战
量身定制,贴合您的项目需求
我们很乐意针对您的具体项目开展定制化评估——基于您的靶点、现有命中化合物或期望的化合物类型。这是在推进下一步之前做出科学、可靠决策的最佳方式。
由计算机辅助药物设计驱动的生物学验证
- 最短化合物交付时间(1-2 天)
- 访问费用享受 25% 折扣
- 定制工作流满足您的需求
- CADD 和生物学服务享受 10% 折扣
- 高概率获得多达 15 个生物学验证的命中化合物(35-75 个工作日内)
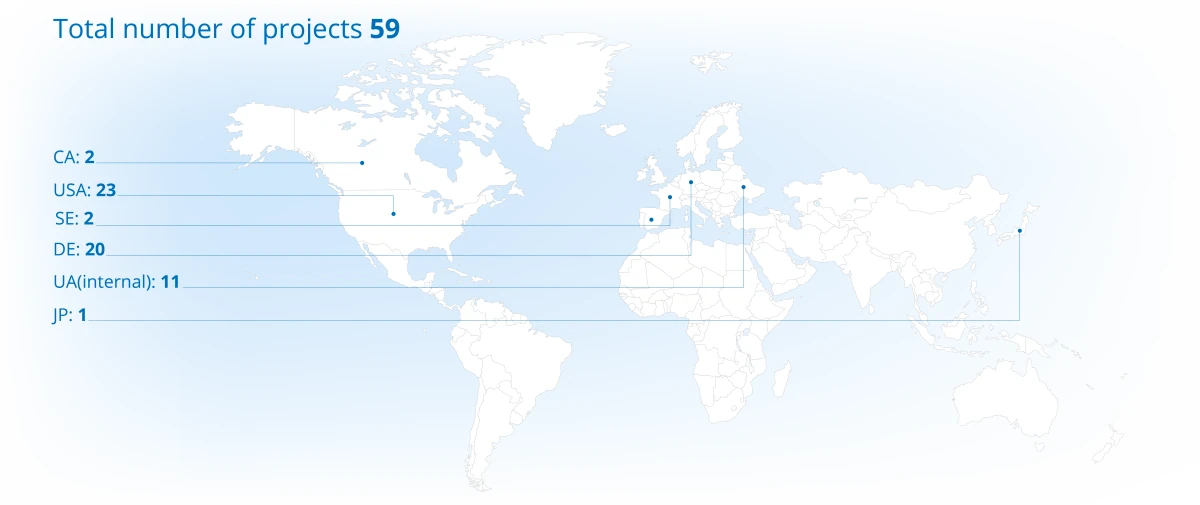
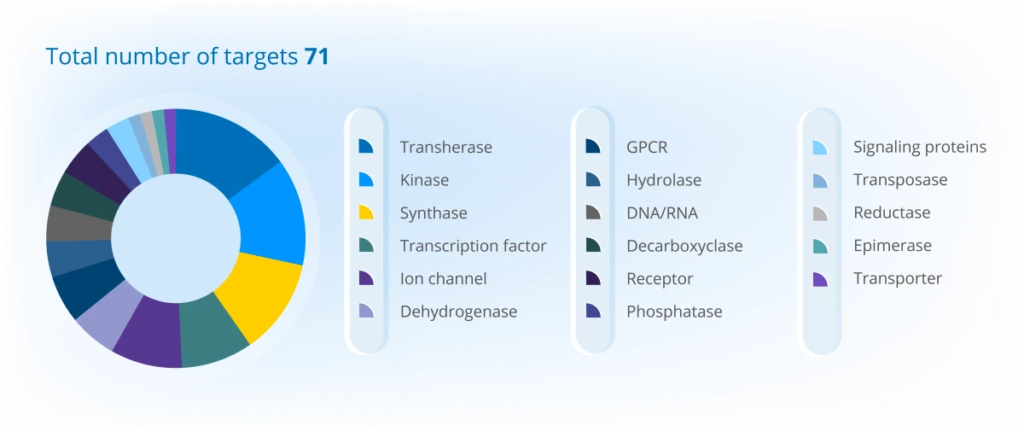
2025 年项目组合概览
项目在各大洲的分布

通过此处的具体案例研究,探索我们在经验和成功方面的深度。
精选的出版物
1. Neural Network Models for Prediction of Biological Activity using Molecular Dynamics Data: A Case of Photoswitchable Peptides
Molecular Informatics 2025, 44. DOI: 10.1002/minf.70001
2. Structural flexibility and shape similarity contribute to exclusive functions of certain ATG8 isoforms in the autophagy process
Molecular Informatics 2025, 44. DOI: 10.1002/minf.70004
3. Pharmacological inhibition of syntenin PDZ2 domain impairs breast cancer cell activities and exosome loading with syndecan and EpCAM cargo
Journal of Extracellular Vesicles 2020, 10. DOI: 10.1002/jev2.12039
4. Structural Insight on the Selectivity of Calyx[4]Arene-Based Inhibitors of Mg2+−Dependent Atp-Hydrolases
Molecular Informatics 2025, 44. DOI: 10.1002/minf.202400200
5. Alternative substrate-assisted hydrolysis pathways of posttransfer editing by prokaryotic leucyl-tRNA synthetase
The FEBS Journal 2025, 292. DOI: 10.1111/febs.70153
6. Lysine Acetylation of Plant α-Tubulins: Scaling Up the Local Effect to Large System Transformations
Proteins: Structure, Function, and Bioinformatics 2025, 93. DOI: 10.1002/prot.26846
7. Computational and experimental identification of putative αTAT1 modulators: implications for nervous system function
Frontiers in Pharmacology 2025, 16. DOI: 10.3389/fphar.2025.1654114
8. Modelling of an autonomous Nav1.5 channel system as a part of in silico pharmacology study
Journal of Molecular Modeling 2021, 27. DOI: 10.1007/s00894-021-04799-w
9. Integrated workflow for the identification of new GABAAR positive allosteric modulators based on the in silico screening with further in vitro validation. Case study using Enamine's stock chemical space
Molecular Informatics 2024, 43. DOI: 10.1002/minf.202300156
10. 4-(Azolyl)-Benzamidines as a Novel Chemotype for ASIC1a Inhibitors
Int. J. Mol. Sci. 2024, 25. DOI: 10.3390/ijms25073584


