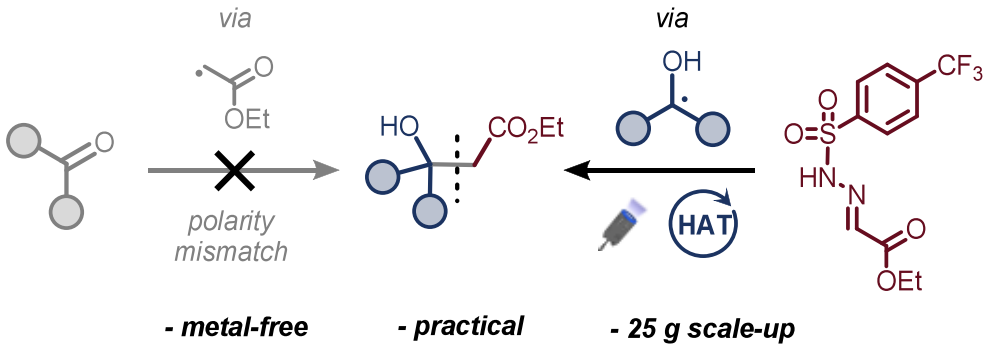
摘要
本文报道了一种市售乙醛酸乙酯磺酰腙试剂,其可通过光催化氢原子转移(HAT)机理与脂肪醇反应,无需依赖过渡金属化学即可生成β-羟基酯化合物。该方案在温和条件下于绿色溶剂中进行,且易于规模化扩展至25克。机理研究表明,初始氢原子转移步骤是光催化转化中的限速步骤,并为碱促进的磺酰肼中间体裂解提供了进一步见解。
自1887年首次报道以来,Reformatsky反应一直是合成有机化学中的核心反应,该反应在锌存在下,通过羰基化合物与α-卤代酯反应生成β-羟基酯(方案1a)。尽管其应用价值已得到证实,但仍存在若干局限性:活泼的溴锌试剂限制了官能团兼容性;有机锌物种的生成通常需要在易燃溶剂(如四氢呋喃、乙醚)中对零价金属进行放热预活化,这在大规模合成中尤其存在安全和重现性问题;此外,该反应还会产生化学计量的金属废弃物,不符合环境和法规标准。β-羟基酯也可通过过渡金属催化的C-H插入反应,由醇与重氮乙酸乙酯偶联制得,但重氮化合物具有潜在爆炸性,阻碍了大规模合成并降低了操作安全性,且需对醇羟基进行保护以抑制固有偏好的O-H插入反应。
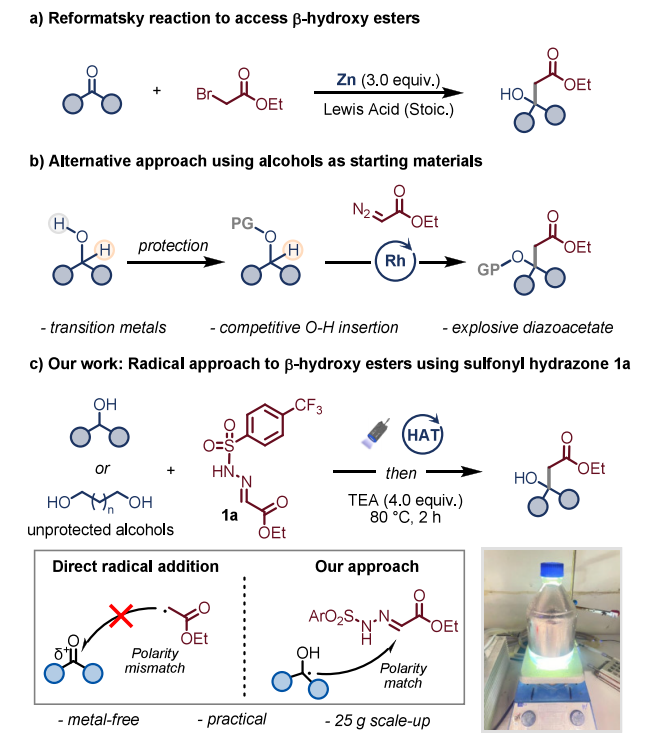
自由基化学的兴起为极性转化中的长期挑战提供了正交解决方案。自由基策略在药物化学中通常具有策略优势,可拓宽官能团耐受性、无需无水条件,并能以羧酸、胺、醇等天然基团作为自由基前体。然而,将这一概念应用于设计形式上的自由基Reformatsky反应仍颇具挑战:亲电α-酯自由基向羰基化合物的直接加成存在根本性极性不匹配问题,导致该过程可逆且热力学上极不利(方案1c)。目前仅报道了局限于醛与亲核三级自由基的反应方法。据我们所知,尚无文献报道无需金属参与的自由基Reformatsky型转化反应,能够实现一级烷基乙酸酯单元的形式上加成。
为解决这些挑战,我们利用了本课题组近期开发且现已市售的乙醛酸乙酯磺酰腙试剂。该稳定试剂可通过脂肪醇的光催化氢原子转移(HAT)途径,与亲核碳中心α-氧自由基高效反应,从而避免了向醛或酮的不利直接加成。该方法采用概念上独特的单电子(1e⁻)策略,以广泛可得的醇作为C-H供体,而非亲电羰基受体。除概念创新性外,该策略使用廉价醇原料降低了成本,并避免了金属活化步骤及金属废弃物的产生。
本文报道了一种无需金属参与的两步法方案,可由脂肪醇和二醇规模化合成β-羟基酯。该转化仅使用易得试剂,在绿色溶剂和工业级溶剂中进行,无需重新优化即可扩展至25克规模。这种由磺酰腙介导的自由基方法不仅为传统极性化学提供了可持续替代方案,还拓展了在温和条件下构建高价值β-羟基酯的合成工具箱。
在光催化氢原子转移步骤的优化中,我们选择环己醇作为C-H供体,与乙醛酸衍生磺酰腙1a反应。采用本课题组先前开发的反应条件(包括光催化偶联及后续磺酰肼中间体裂解),经两步反应以69%的核磁共振产率获得了目标β-羟基酯4h(表1,条目2)。将溶剂更换为乙醇后,在过量三乙胺存在下,磺酰肼3h的裂解可高效进行,2小时内即可定量脱除活化基团。通过对不同光催化剂、溶剂和反应条件的系统筛选,我们发现以二苯甲酮作为氢原子转移光催化剂、无水丙酮(0.2M)作为溶剂时,产率可提升至93%(表1,条目1)。有趣的是,未优化条件下,该反应在水相(0.1M)或无溶剂体系中仍表现出良好反应性,产率分别为50%和79%(表1,条目3和4)。初步实验表明,一级醇的反应性低于模型体系,我们尝试筛选多种路易斯酸作为添加剂,但未观察到产率改善(见支持信息第5节)。对乙醛酸乙酯磺酰腙的电子效应研究表明,最初开发的4-三氟甲基取代衍生物1a在本反应条件下性能最佳(表1,条目5和6)。
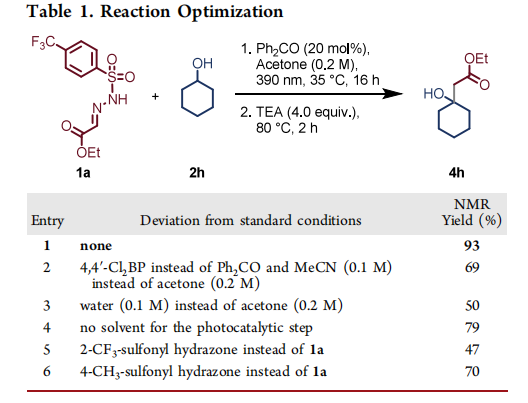
通过在裂解步骤中直接用乙醇稀释乙酸乙酯反应液,该方案成功适配为一锅法流程,且产率无显著损失(见支持信息第5节)。但为保证实验一致性和更广适用性,底物范围考察仍采用两步法方案。
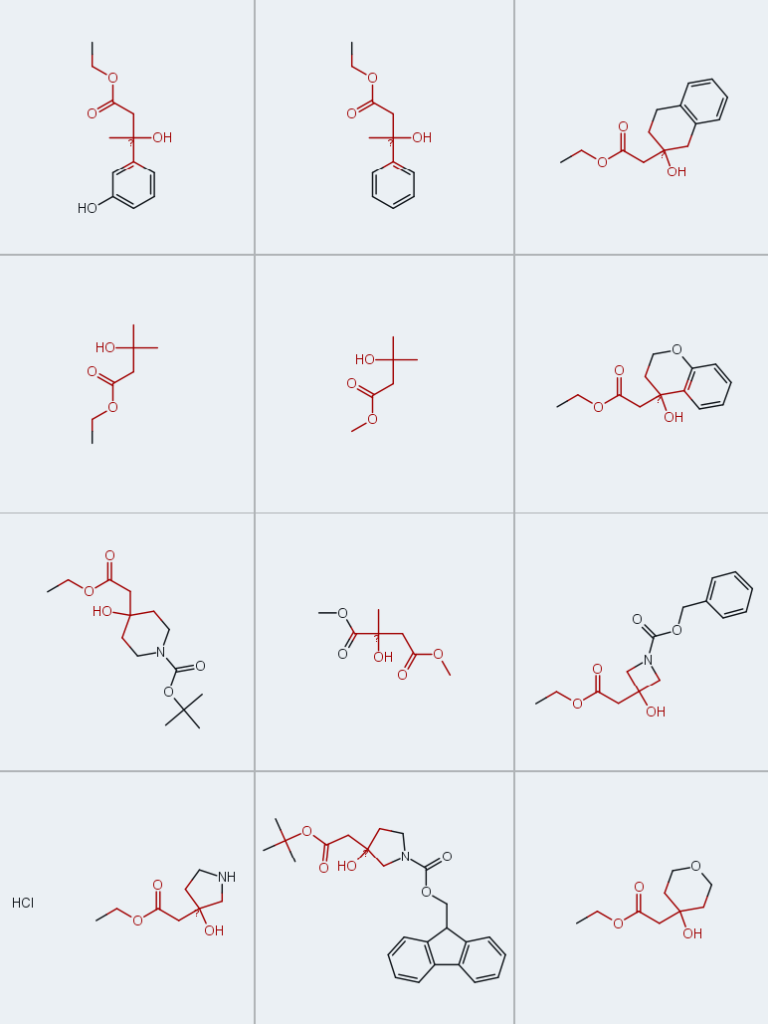
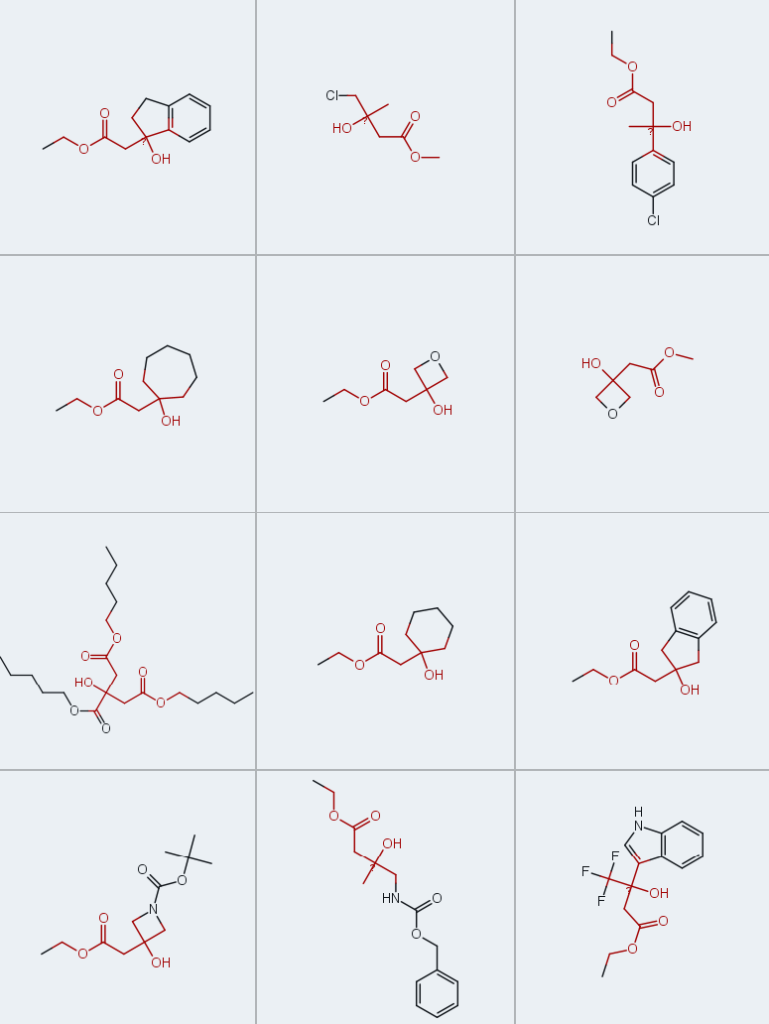
在优化条件下,我们考察了该转化的普适性(方案2)。尽管反应性较低,一级醇在该方案中仍表现良好,以中等产率生成化合物4b−4e。值得注意的是,乙醇(2a)可适用于该方法,中间体3a的分离产率达56%;环状二级醇反应性优异,化合物4f−4k的产率为54%−87%;含酯基(4l、4q)、芳环(4n)、氯原子(4e)、三氟甲基(4d)和叔丁基二甲基硅基保护基(4t)的醇均能耐受该反应条件,获得中等到良好产率。尤为突出的是,不同链长的未保护二醇与该方法具有良好兼容性,化合物4r、4s和4u的分离产率为36%−77%。二醇的反应性具有重要意义,其应用为传统Reformatsky反应提供了互补方法,拓宽了可获得的β-羟基酯范围,这种灵活性在化合物库构建和组合化学应用中尤为宝贵。
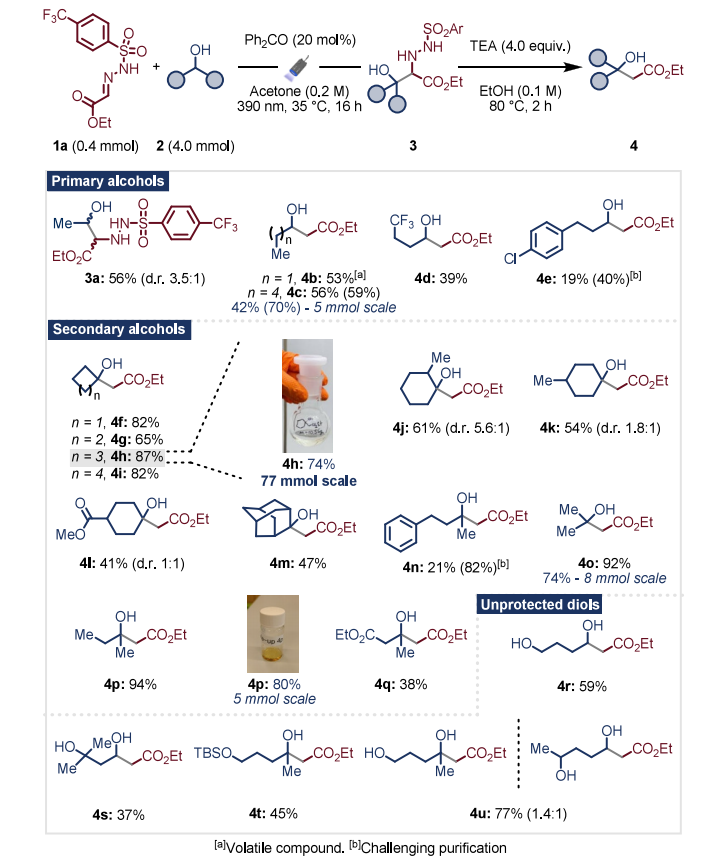
我们通过β-羟基酯部分的后期官能化进一步证明了该方案的实用性,可快速获得具有多样官能团的高价值合成砌块(方案3a):通过经典的里特型反应制备4-甲基苯甲酰胺衍生物5;酯基完全还原以79%产率得到二醇6;化合物4o经酯水解后与吗啉酰胺化,两步反应以83% 产率生成酰胺8。
该方法在规模化合成中表现出良好的稳健性:5毫摩尔规模制备化合物4c时,两步反应的核磁共振产率达70%,高于0.4毫摩尔规模的59%;8毫摩尔规模制备4o和5毫摩尔规模制备4p时,产率分别为74%和80%,与小试规模相当;模型底物4h无需进一步优化即可扩展至25克规模,分离产率为74%(10.5克)。

我们进行了机理研究以验证方案3b中提出的光催化循环(见支持信息第7节,机理研究)。对单个组分及完整反应混合物的紫外-可见吸收光谱分析表明,二苯甲酮是390nm处吸收的主要贡献者。观察到的动力学同位素效应(KIE=3.1)、反应速率对二苯甲酮负载量的强依赖性以及对总反应浓度的弱依赖性,表明生成α-氧自由基II的初始氢原子转移步骤是催化循环中的限速步骤。量子产率测定(Φ=0.06)支持这一结论,并排除了自由基链反应的参与(方案3b)。
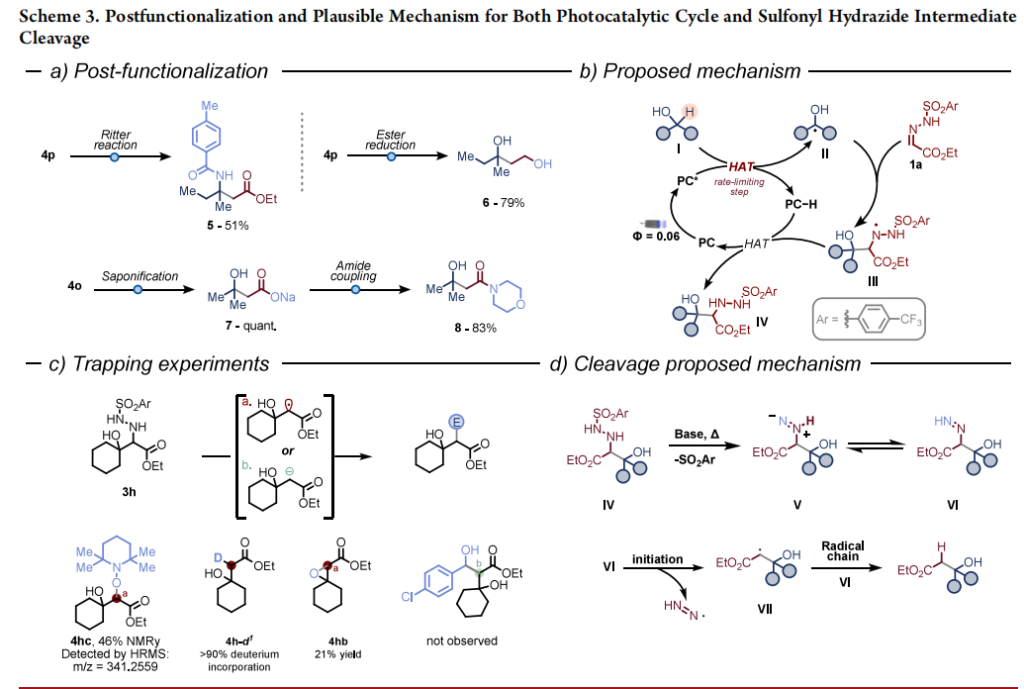
阐明光催化步骤机理后,我们进一步研究了碱介导裂解过程的本质(方案3c):向反应体系中加入2,2,6,6-四甲基哌啶-1-氧基(TEMPO)可显著抑制反应,通过高分辨质谱(HRMS)检测到TEMPO自由基加合物4hc,并经¹H核磁共振定量;使用四氯化碳进行自由基捕获实验,经溴化和后续分子内亲核取代反应生成环氧化物4hb(产率21%),同样表明裂解过程具有自由基特性;而加入氯苯甲醛和苄基溴未观察到捕获产物,排除了阴离子途径。氘代实验结果与卢等人事先在2024年报道的结果一致,进一步支持自由基裂解途径(方案3c及支持信息第7.9节)。基于上述实验证据和文献报道,我们提出了合理的反应机理(方案3d):肼中间体IV的酸性质子首先去质子化,引发亚磺酸盐离去并生成异重氮烯中间体V,V与重氮烯VI处于平衡状态;随后自由基引发生成关键自由基中间体VII;通过与新的重氮烯VI分子发生氢夺取的自由基链机理,最终生成β- 羟基酯产物。
综上所述,本文报道了一种无需金属参与的光催化形式上自由基Reformatsky转化反应,该反应通过易得醇与亲电乙醛酸乙酯磺酰腙的极性匹配C-C键形成,实现了高价值β-羟基酯的高效合成。该方法仅使用市售试剂和绿色溶剂,操作简便,且易于扩展至至少25克规模。这些特性提升了方案的实用性,并降低了工业应用中的安全和法规顾虑。此外,机理研究为芳基磺酰腙和磺酰肼的自由基反应性提供了宝贵见解,拓宽了其在合成方法学中的应用潜力。
Enamine始终深耕新型砌块分子的研发领域,针对本文聚焦的药物化学关键中间体——β-羟基酯,现已储备近百种现货产品,可随时供全球客户选用: