
摘要
将金(I)催化的脂肪族1-溴代炔烃环异构化反应应用于杂螺环化合物的合成中。产物中碳(sp²)-溴键的反应活性使得所获得的骨架能够进一步衍生化。通过这种方式,可轻松得到带有多种官能团(如羧基(CO₂H)、氨基(NH₂)、羟基(OH)和频哪醇硼酸酯基(Bpin))的螺杂环化合物。这些螺环化合物是现代药物研发项目中极具价值的砌块。
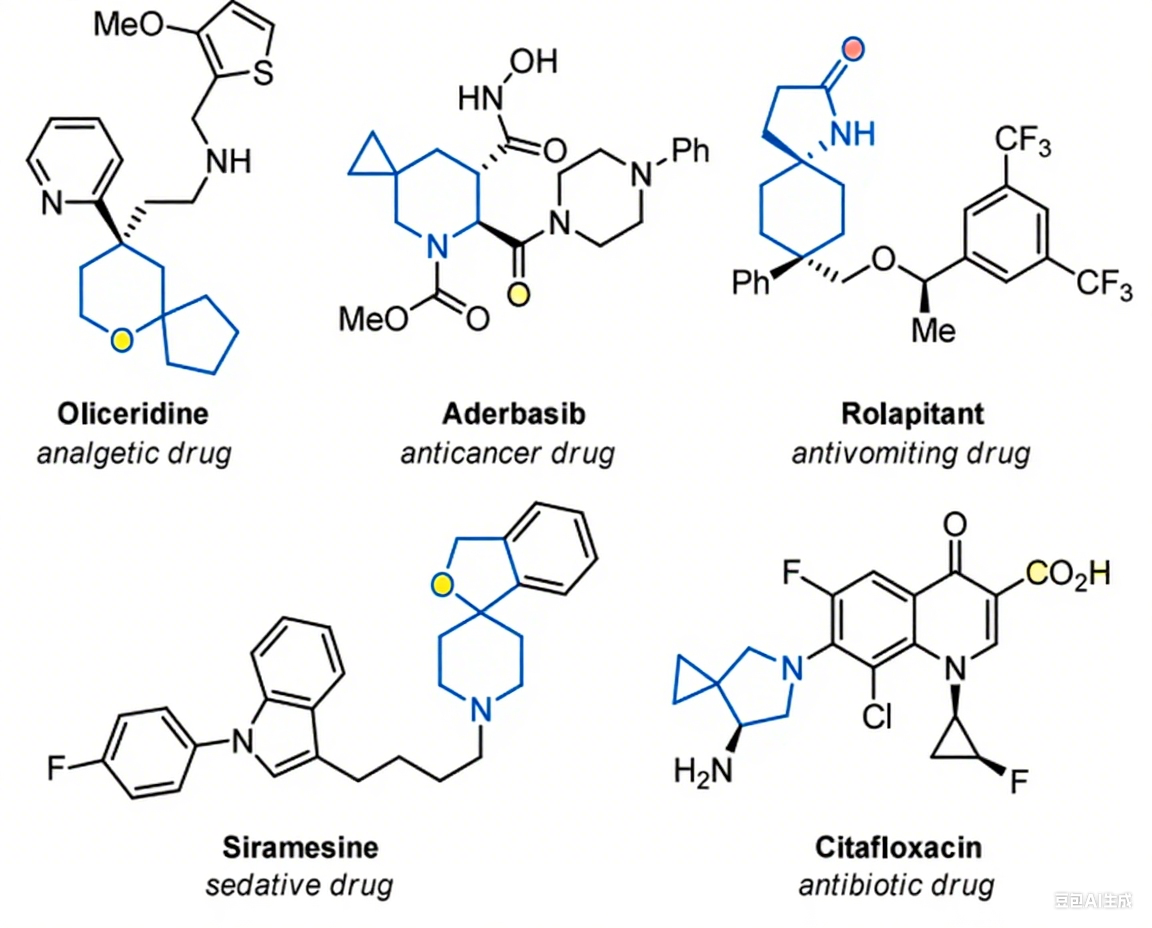
含杂原子的螺环化合物在现代药物研发的发展进程中发挥着关键作用 [1]。这类结构单元不仅广泛存在于具有生物活性的分子中,还是多种临床获批药物的重要组成部分(图 1)。它们独特的化学性质和药代动力学性质通常能转化为更强的结合亲和力、选择性和代谢稳定性,使其成为药物化学中极具价值的骨架结构。鉴于其重要性,开发新的高效合成方法来构建这类骨架结构始终是研究的重点方向。其中,氧杂螺环化合物和氮杂螺环化合物尤为受关注。
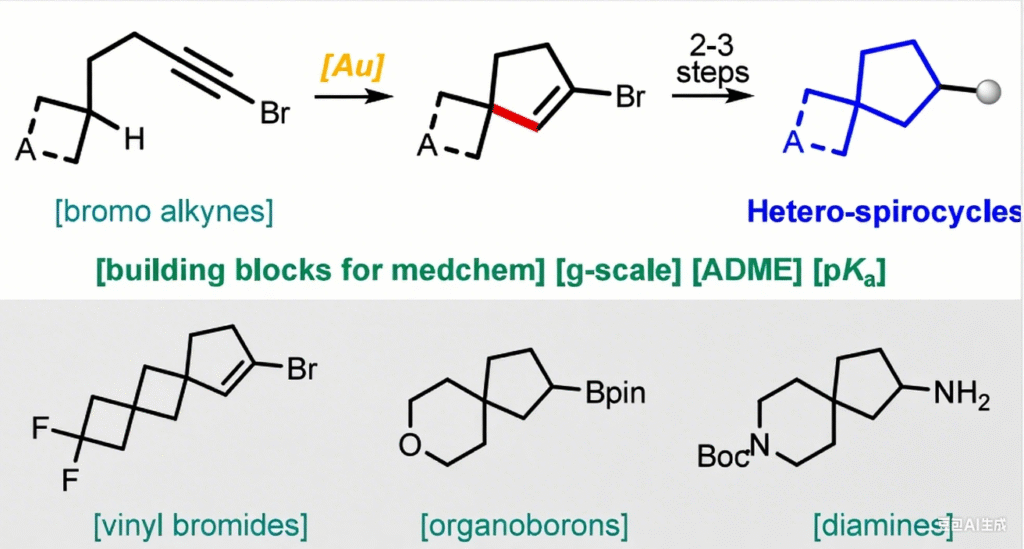
含杂原子的螺环化合物,尤其是含有氮、氧等原子的螺环化合物,凭借其刚性的三维结构以及在改善类药性方面的潜力,受到了越来越多的关注 [2,3]。在此背景下,我们提出了一种新颖且实用的方法,通过金(I)催化 [4] 脂肪族 1-溴代炔烃 [5,6] 的环异构化反应来合成氧杂/氮杂螺环化合物。该策略在温和条件下为获取结构多样的螺环体系提供了一条简便高效的途径,丰富了构建这类重要结构单元的合成方法库。
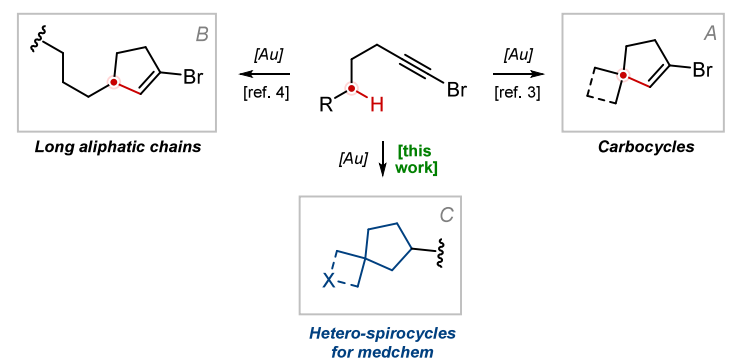
近期,我们报道了一种新型金(I)催化的脂肪族 1-溴代炔烃环异构化反应,该反应实现了未活化 C (sp³)−H 键的官能团化 [7-9]。从骨架可及性来看,该反应展现出优异的普适性:除简单环戊烯衍生物外,通过在起始原料中引入碳环,还可得到桥环、并环和螺环双环骨架(图 1A)。然而,该反应的主要局限在于官能团耐受性较窄。在后续研究中我们发现,对于线性底物,若要使反应顺利进行,官能团与溴原子之间至少需间隔 7 个碳原子(图 1B 和图 2)[10]。本研究中,我们报道了少数特定杂环底物同样可参与该反应,进而为药物化学领域提供高附加值的杂螺环砌块(图 1C)。
初步研究结果
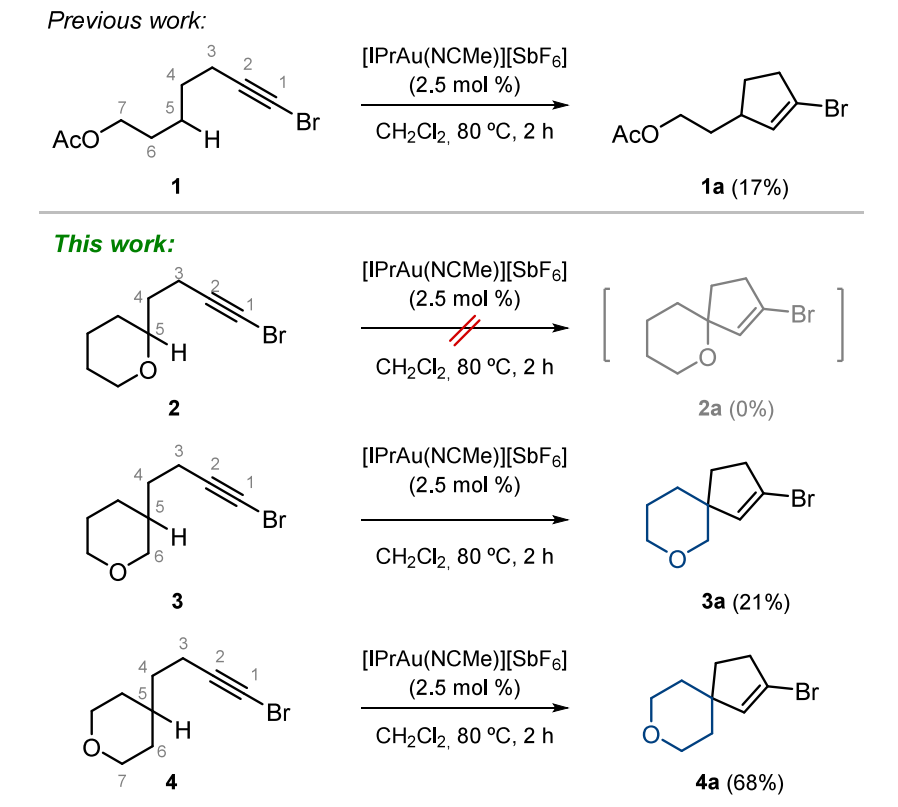
此前,我们证实化合物 1 能够发生金(I)催化的环异构化反应,生成乙烯基溴化物 1a,但产率较低,仅为17%(见表 2)[9]。受此发现的启发,我们推测含环状氧原子的底物可能也会发生类似的环异构化反应。然而,在该反应条件下,化合物 2 未发生任何变化并得以回收。相反,化合物 3 生成了目标产物,尽管产率较低,但仍达到了 21%,这一结果令人鼓舞(见表 2)。环状底物的这一反应结果与我们对直链类似物的初步研究发现相符,表明只有当杂原子与溴原子之间的距离足够远时,底物才能发生环异构化反应。不过,在这类环状系列化合物中,反应活性受与反应中心距离的影响似乎更小,这一趋势逐渐显现。
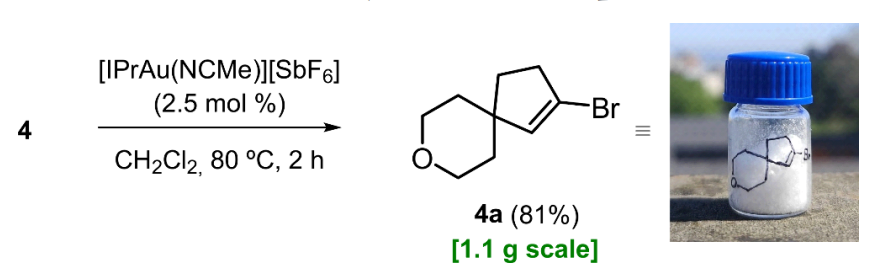
在直链系列底物中,杂原子必须至少位于第 7 位;而当杂原子嵌入杂环、处于第 6 位时,我们观察到了相近的产率。我们认为,这一位置差异(带来的产率变化)归因于底物环状结构所施加的构象限制。最显著的结果是在异构体溴代炔烃 4 的反应中得到的,该反应生成目标螺环产物 4a 的产率大幅提高,达到了 68%(见表 2)。再次证明,产率的提高体现了上述杂原子对距离的耐受性。
适用范围
这些初步研究结果为探究该合成策略的适用范围和局限性奠定了基础;本研究仅考虑远程螺环化反应。首先,除了吡喃 4a 之外,我们还对带有其他含氧官能团(如缩酮 5a 或酮 6a)的六元碳环化合物进行了测试。在证实该反应对多种含氧官能团具有兼容性后,我们将研究重点转向了氮杂环化合物。在这种情况下,需要考虑一个额外的变量,即氮原子上的保护基。我们对多种保护基(7 - 9)进行了筛选,结果发现三氟乙酰胺 7a 的反应效果最佳。随后,我们研究了在 4 位带有其他药效团的六元环化合物,如二氟代化合物 10a、砜类化合物 11a 和偕二甲基化合物 12a。二氟亚甲基和偕二甲基单元的反应产率处于中等至较高水平(10a 和 12a;表 4),而砜衍生物 11a 的产率较低,仅为 13%。为了验证 “溴代炔烃与杂原子的相对位置是决定反应活性的关键因素” 这一假设,我们决定将环的尺寸从六元环扩大到七元环。氧杂卓衍生物 13a 的反应产率为中等水平(32%)。令人满意的是,相应的七元环内酯生成产物 14a 的产率更高,达到了 76%。随后,我们设想使用螺 [3.3] 庚烷衍生的溴代炔烃可能会得到类似的结果,因为溴代炔烃与极性基团的相对位置能够保持不变。正是这一特性使得这类骨架结构可用作六元杂环化合物的生物电子等排体 [11]。2,2-二氟螺[3.3]庚烷 15 生成目标产物 15a 的产率(74%)高于六元环类似物 10a(50%)(表 4)。在溴代炔烃单元与螺环之间的连接链中引入碳环,能够生成双螺环化合物 16a,产率为中等水平(51%)。在这种情况下,需要对反应条件进行轻微调整(将反应温度从 80℃降至 60℃,反应时间从 2 小时延长至 12 小时),以最大限度地减少副产物的生成。最后,我们使用多种三元取代环化合物,对吡喃环反应中观察到的规律的普遍性进行了初步评估(见表 2)。令人欣喜的是,氮杂卓、酮和偕二氟衍生物 17a - 19a 的产率处于中等至较高水平。这些结果进一步证实了上述观点,即在环状体系中,杂原子类官能团可以在距离反应中心更近的位置存在,且不会显著影响反应。
局限性
我们发现该合成方法主要存在三个局限性(表 4 底部及支持信息)。首先,N-叔丁氧羰基(Boc)保护基不适用。在反应中,仅检测到分子间亲核加成产物 20b,且其产率较低,仅为 18%[12]。其次,如前所述,当杂原子距离反应中心过近时,较小的环(氮杂环丁烷 38a 和四氢吡喃 41a;见支持信息)或在连接链中带有杂原子的底物(39a;见支持信息)无法发生反应。基于上述相同的思路,我们尝试使用 2-氧杂螺 [3.3] 庚烷衍生的底物作为吡喃环的电子等排体。但遗憾的是,由于起始原料在反应条件下发生分解,未能得到相应的衍生物 40a(支持信息)。
修饰
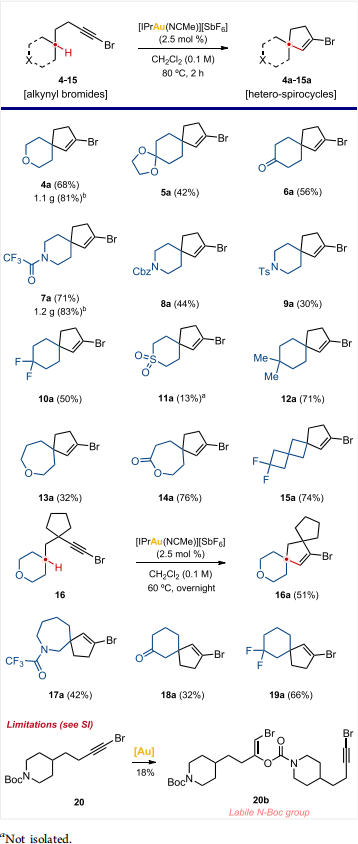
明确了金催化螺环化反应的适用范围和局限性后,我们进一步研究了合成产物中存在的具有多功能反应活性的碳(sp²)- 溴键的反应性能(表 5)。在众多可能的反应中,我们选择了那些能够生成带有药物研发中常用官能团(如羧基、氨基、酮基或硼酸酯基)的衍生物的反应(表 5)。在这项衍生化研究中,我们仅使用了氧杂和氮杂衍生物 4a、7a 和 20a。通过羧基化反应,随后进行催化氢化,得到了羧酸衍生物 21a/21b(表 5)。羧基的多功能性使其能够进一步反应生成相应的醇 22a 以及两类不同的氨基衍生物 23a/23b 和 24a/24b,这两种氨基衍生物可分别通过酰胺还原反应或Curtius重排反应制得。在Arndt−Eistert条件下,还能得到同系化的羧酸 25a。最后,通过Suzuki硼酸化反应,随后进行催化氢化,得到了硼酸酯衍生物 27a(表 5)。碳-硼键的氧化反应能够生成酮 28a/28b,由酮 28a/28b 可轻松合成三氟甲基醇 29a。
官能团的酸性
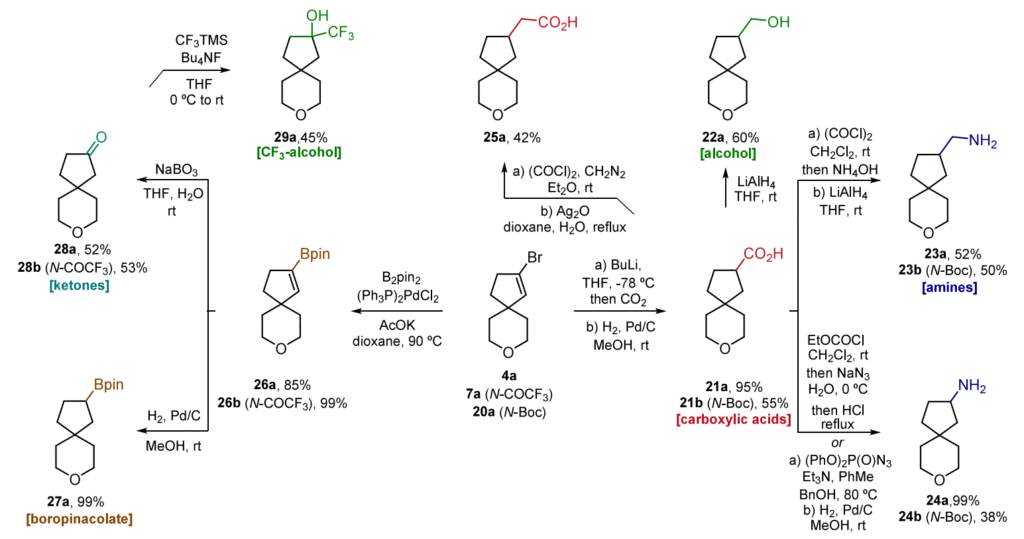
我们还研究了用螺环类似物取代吡喃环对化合物电子性质的影响。为此,我们通过实验测定了羧酸 21a 和 28 以及氨基盐酸盐 24a 和 29 的 pKa 值(见图 2)。含有吡喃环的羧酸 28 的酸性比螺环羧酸 21a 强 0.5 个 pKa 单位:28 的 pKa 值为 4.2,而 21a 的 pKa 值为 4.7。在氨基化合物中也观察到了类似的趋势。含有吡喃环的氨基盐酸盐 29 的酸性比螺环氨基化合物 24a 强 0.8 个 pKa 单位:29・HCl 的 pKa 值为 9.7,而 21a・HCl 的 pKa 值为 10.5。
物理化学性质和药代动力学性质
最后,我们希望了解将单环化合物替换为螺环化合物对有机化合物实验物理化学性质和药代动力学性质(水溶性、脂溶性和代谢稳定性)的影响。为此,我们合成并研究了四种模型酰胺化合物 30 - 33。由于这四种化合物的溶解度过高且稳定性过强,超出了实验方法的检测灵敏度范围,因此未观察到将吡喃环替换为螺环类似物对实验水溶性(sol.)和代谢稳定性(CLint)产生影响(图 3)。将吡喃环替换为更大的螺环类似物后,化合物的脂溶性增加了约 1 个 logP 单位:30 的 logP 值为 1.4,31 的 logP 值为 2.4;32 的 logP 值为 1.1,33 的 logP 值为 2.0(图 3)。
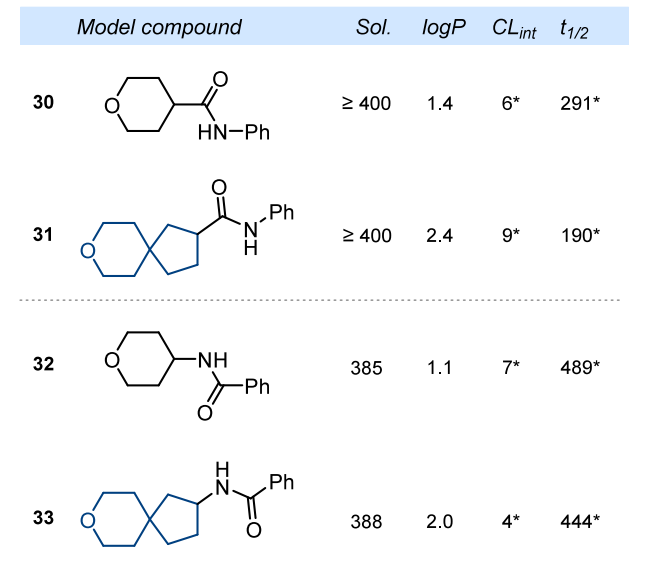
注:溶解度(Sol.):在 pH 值为 7.4 的磷酸盐缓冲液中的实验动力学溶解度(单位:μM);脂溶性(logP):在正辛醇 - 水体系中的实验分配系数,可靠的 logP 值范围为 1.0 - 4.0;内在清除率(CLint):在人肝微粒体中的实验代谢稳定性(单位:μL・min⁻¹・mg⁻¹);半衰期(t₁/₂):代谢分解的实验半衰期(单位:min)。(*)由于化合物稳定性极高,该参数仅为近似值。
综上所述,本研究开发了一种通过金(I)催化独特的杂环 1-溴代炔烃环异构化反应来合成杂螺环化合物的新颖实用方法。与直链底物相比,环状底物对距离较近的杂原子具有更好的耐受性。产物中存在的易于官能团化的碳(sp²)- 溴键被充分利用,用于引入多种常见的官能团,如羧基(CO₂H)、氨基(NH₂)、羟基(OH)和频哪醇硼酸酯基(Bpin)。所获得的杂螺环衍生物是药物研发项目中极具价值的构建模块。目前,研究人员正在进一步探索更具耐受性的反应条件,以扩大该方法可合成的杂螺环化合物的范围。