
编者按:上一期,我们介绍了 Enamine 团队发表于《Chemistry—A European Journal》的研究成果,该论文提出一种基于 Petasis/Grubbs 反应序列的类多样性导向合成(DOS)策略,可用于制备螺环类药物化学相关砌块。本期我们将继续分享 Enamine 团队的另一项研究——发表于《The Journal of Organic Chemistry》的论文,该研究同样以 Petasis/Grubbs 反应序列为核心,实现了δ,δ-螺取代δ-氨基酸及其 α,β-不饱和类似物的高效合成
摘要
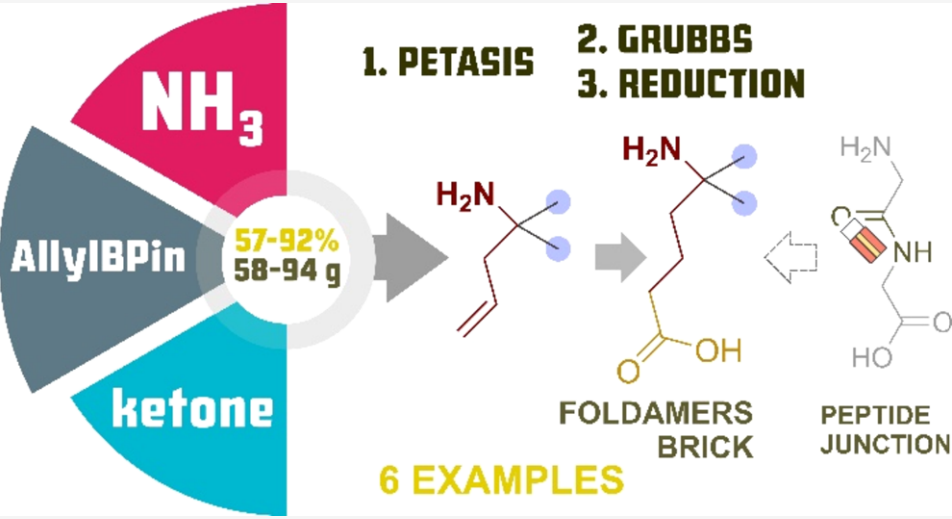
本文开发了一种从大宗酮类化合物合成 δ,δ-螺取代 δ-氨基酸及其α,β-不饱和类似物的便捷可扩展方法。所提出的路线包括起始酮与烯丙基硼酸频哪醇酯和甲醇氨的 Petasis 反应,以及与丙烯酸衍生物的交叉复分解反应作为关键步骤。所开发的方案新颖、稳健且经济高效。它们避免了繁琐的分离和纯化步骤,可放大至40克最终化合物规模,并为新型构象刚性折叠体提供了高效合成途径。
引言
在所有双功能砌块中,氨基酸(AA)是有机合成和药物化学中最知名且应用最广泛的一类。这类化合物之所以占据重要地位,原因如下:(i)是所有已知生物体的组成成分;(ii)是手性池中的常用砌块;(iii)是药物发现中的重要试剂,具有药物化学家常用的最常见官能团;(iv)可调节有机分子中肽的性质。除了 20 种经典氨基酸外,大量非经典和非天然氨基酸被用于现代药物化学中。设计非天然氨基酸的经典方法通常包括构象限制、氟化和/或等排体替换。多年来,我们在设计中经常使用上述方法,但通常我们的合成仅限于 α、β 和 γ 型氨基酸。最近,我们也将注意力转向了 δ- 氨基酸。这类氨基酸是折叠体的重要砌块、复杂哌啶(包括天然哌啶)的前体,以及现代药物发现的砌块。在含此类片段的研究药物中,诺和诺德开发的口服生长激素 secretagogue(GHS)他莫瑞林(NN-703)及其类似物值得关注(图 1A)。此外,由于未受限制的母体衍生物具有较大的柔性,δ- 氨基酸链的构象锁定对于新型药物折叠体的设计至关重要。这可以通过在氨基酸核心中添加双键、稠环或螺环来实现。类比于 α-和 β-氨基酸在肽中的螺环锁定,我们决定开发一种实用的方法来合成用于折叠体的 δ,δ-螺取代氨基酸。这类化合物此前已有报道,但仅通过难以实现的方法合成。
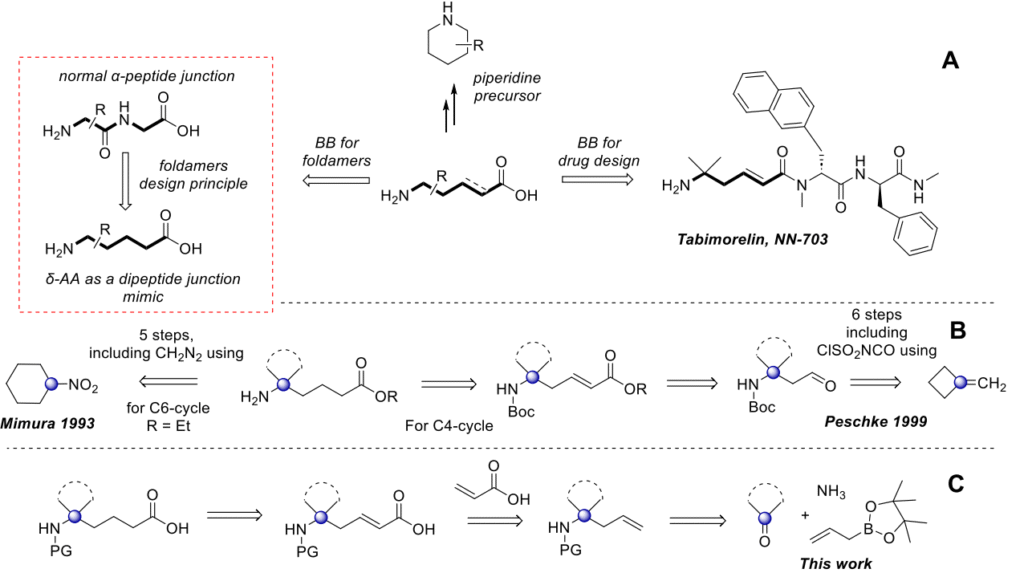
第一种方法针对含螺环己基的衍生物,通过对不便使用的硝基环己烷进行五步修饰实现,包括基于 CH₂N₂的链延长步骤。另一种方法基于相应β-氨基酸衍生物的C2延长,该衍生物通过以亚甲基环丁烷为起始原料、利用与 ClSO₂NCO 的 [2+2]环加成作为关键步骤的六步序列获得。这两种方法都不适用于放大生产,且对最终δ-氨基酸设计中适用的环多样性有限。
本文报道了我们开发的从易得大宗酮类化合物经四步序列合成 δ,δ-螺取代 N-保护 δ-氨基酸的方法。该序列包括高效可扩展的反应:与氨和烯丙基硼酸频哪醇酯的 Petasis 反应、适当的胺保护、与丙烯酸的交叉复分解反应以及催化氢化。本研究是我们类多样性导向合成(DOS)项目(利用 Petasis/Grubbs 组合设计和制备合成药物化学相关砌块)的一个原创且有前景的分支。
在本研究中,我们选择了一组大宗商业酮类化合物,如方案 1 所示。其中包括最简单的无环丙酮 1a、C4-C6 环酮 1b-d、吡喃酮 1e 和 N-Boc 保护的吡咯烷酮 1f。所有这些酮都通过与我们之前开发的方案类似的方法,成功与烯丙基硼酸频哪醇酯和甲醇氨溶液进行 Petasis 反应,以 57-92% 的产率得到目标高烯丙基胺 2。值得注意的是,这六种化合物中的三种(2a、b、e)优选以盐酸盐形式分离(见支持信息(SI))。在所有情况下(游离胺或相应盐),合成过程均避免了色谱纯化,并成功放大至约 100 克最终产物规模。放大的成功使我们能够将这些胺视为进一步多样化设计和合成药物化学相关砌块的良好起点。胺 2a-e 用 Boc₂O 在二氯甲烷中进行 Boc 保护,对于盐酸盐(化合物 2a、b、e)使用三乙胺作为碱,对于 1c、d 则无需碱,以 93% 至定量的产率得到保护形式 3a-e,规模约 100 克,同样避免了色谱纯化(步骤 b)。下一步是与丙烯酸甲酯的交叉复分解反应。尽管有报道称此类反应需要 CuI 催化,且最近已用于大规模反应,但我们发现使用第二代 Grubbs 催化剂在二氯甲烷中的经典无铜条件效果良好。结果,交叉复分解反应被放大至约 50 克最终产物 4a-e,制备产率为 76-85%(步骤 c)。纯化过程包括将反应混合物通过滤纸上的硅胶层(或短柱)过滤,使用己烷 / 甲基叔丁基醚 4:1 作为洗脱剂。在该体系中,化合物 4a-e 的 Rf 值在 0.4-0.5 范围内,这使我们能够使用快速色谱法获得纯产物,且均为反式异构体。化合物 4a-e 在无水乙酸乙酯中,在 10% Pd/C 存在下,于 1atm 氢气下进一步催化氢化,通过简单过滤除去催化剂并真空蒸发溶剂,得到相应化合物 5a-e,产率几乎定量(步骤 d)。化合物 5 型的水解通过在甲醇溶液中用氢氧化钠水溶液实现(步骤 e)。通过连续蒸发/萃取程序(见支持信息),在单次合成运行中以 20 至 40 克规模得到最终目标 N-Boc 保护的酸 6a-e,产率 90-99%。
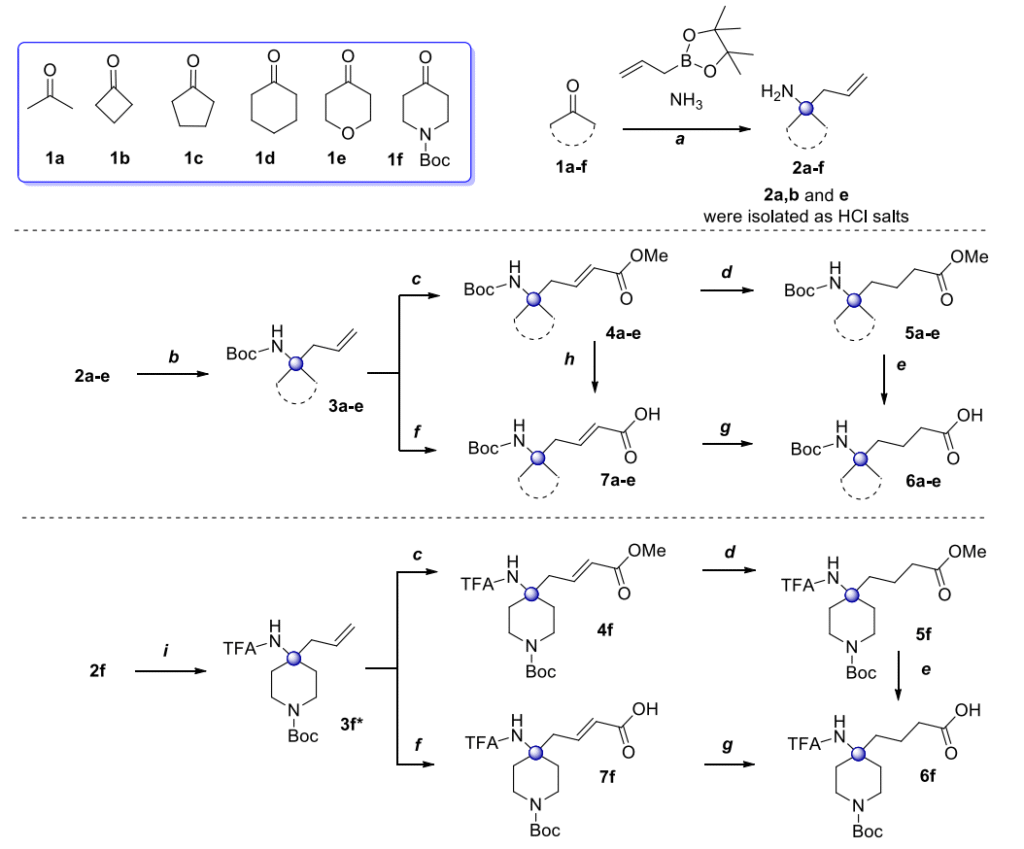
相应的不饱和氨基酸 7 型作为砌块也很有意义(例如,化合物 7a 是他莫瑞林类似物的前体)。然而,由于迈克尔加成副反应,在碱性条件下(步骤 h)水解丙烯酸存在问题。为避免该问题,我们测试了在反应中使用游离丙烯酸代替酯。在上述条件下使用市售丙烯酸样品导致转化率极低,这更可能是由于存在难以检测的缩聚杂质。尽管如此,我们还是找到了一个原创解决方案:将市售样品冷冻至 + 4°C,可使高纯度丙烯酸结晶。在上述温度下收集晶体后,立即用于反应,在沸腾的二氯甲烷中 2-8 小时内可实现完全转化(见 SI)。我们还发现,在这种情况下,交叉复分解反应对 3 型化合物的纯度更敏感。即使根据 NMR 判断它们具有足够的纯度,在复分解步骤之前将起始烯烃通过硅胶(己烷/甲基叔丁基醚 10:1 作为洗脱剂)过滤也至关重要,这显著提高了反应速率并减少了所需催化剂的量。在这些条件下,处理后通过硅胶柱色谱(己烷/乙酸乙酯 2:1 作为洗脱剂),最终产物 7 的制备产率在 63-69% 范围内,规模为 20-25 克(步骤 f)。值得注意的是,7 型酸也可以催化氢化为6型酸(步骤 g),但尽管步骤数减少,这种方法的效率较低。因此,化合物6d的总产率(步骤 c-d-e)达到 67%,而(步骤f-g)为 52%。
遵循上述步骤序列,带有两个正交保护基(TFA 和 Boc)的化合物 2f 在不改变为 3a-e 开发的一般方案的情况下得到了类似结果。值得注意的是,在步骤 e 中,N-TFA 保护对酯的碱性水解具有耐受性。不同步骤的具体产率取决于起始酮的结构,汇总数据列于表 1 中。
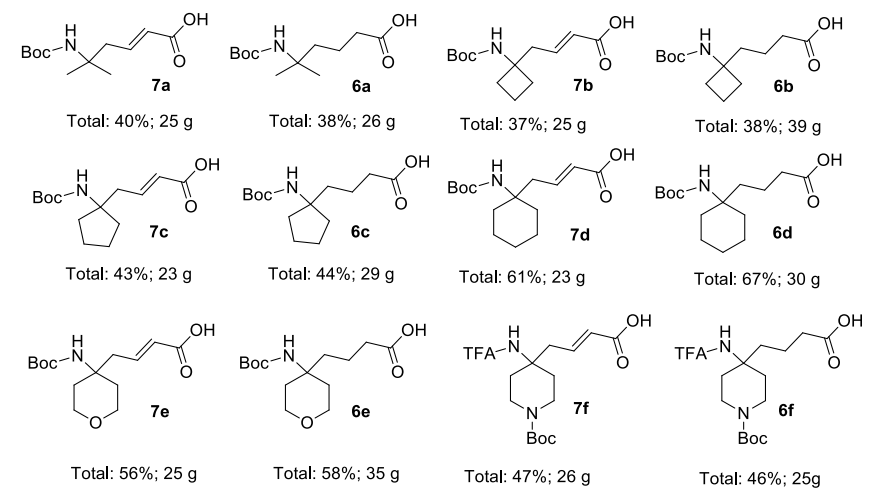
最后,开发了一种从大宗酮类化合物合成 δ,δ-二取代 δ-氨基酸及其不饱和类似物的全新路线。结果,合成了 12 种氨基酸的小型库(图 2),单次合成运行的量高达 40 克。所有程序均设计为避免繁琐的纯化操作。关键的合成创新基于数百克规模的 Petasis 步骤放大和交叉复分解反应的重要改进,特别是两种组分纯度的重要性。所提出的方案易于放大,可用于生产药物化学砌块和 API 合成的高级中间体。展示了所用酮的多样性以及该方案对 Boc 和 TFA 氮保护的耐受性。因此,所提出的方法有望通过使用各种市售起始材料显著扩展有形化学空间。