
编者按:这篇由Enamine发表于《Chemistry—A European Journal》的论文,提出一种基 Petasis/Grubbs 反应序列的类多样性导向合成(DOS)策略,用于制备螺环类药物化学相关砌块。研究优化了 Petasis 反应条件,结合 Grubbs 关环复分解反应,在克级规模合成出 16 种基于螺环哌啶的新型 Bemis-Murco 骨架。所得砌块包括单功能、双功能和三功能类型,符合相关数据库要求。化学信息学分析显示,这些模块能显著拓展化学空间,生成的化合物新颖性高,为药物发现提供了高效途径。
通过这项模型研究,我们展示了在砌块层面运用类多样性导向合成(DOS)方法,结合成熟的平行合成方法进行修饰和拓展超大有形化学空间,以提高药物发现筛选效率和质量的广阔前景。该策略使我们得到了 16 种基于功能化螺环哌啶的新型 Bemis-Murco 骨架。通过多克级规模的 Petasis/Grubbs 反应序列,实现了上述结构单元的高效、可制备且可扩展的类 DOS 合成方法。化学信息学生成的酰胺类化合物库显著拓展了有形化合物的化学空间(以 Enamine 的 REAL 数据库为例)。
1. 引言
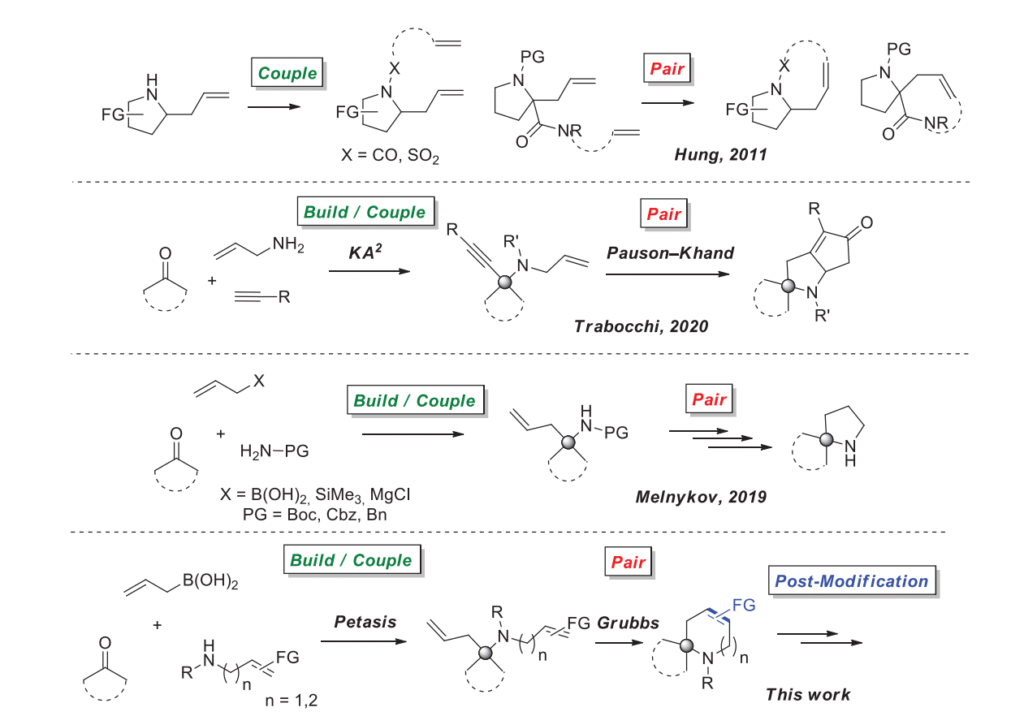
过去二十年来,药物化学对现代有机化学产生了深远影响。这一点从 DOS、导向合成(LOS)和 DNA 模板合成(DTS)等新概念的出现就可清晰看出,这些概念是经典有机合成方法的补充 [1]。DOS 概念最初的流行随着时间的推移受到了诸多批评,因为它会产生仅在化学遗传学中有用的 “肥胖” 分子 [2]。作为解决方案之一,从 Hung 的开创性研究开始,DOS 方法开始被用于设计 “小型” 分子,包括用于基于片段的药物设计(FBDD)[3,4]。另一种用于螺环化合物的有前景的 DOS 方法基于多组分反应,通过环化步骤得到目标加合物。在众多实例中,多组分酮 - 胺 - 炔(KA2)偶联与 Pauson-Khand 反应的结合值得关注 [5]。受这些研究的启发,我们决定尝试将 DOS 概念应用于更具挑战性的、带有螺环部分的药物化学相关砌块的设计任务中。螺环片段在现代药物化学和药物发现中广泛存在。例如,螺环胺及其衍生物在药物化学中被广泛用于治疗各种疾病,从中耳炎、丙型肝炎到精神分裂症和焦虑症 [6]。因此,我们选择模型反应来证明将 DOS 概念用于砌块设计的可行性,这一选择是顺理成章的。在众多多组分反应中,我们将注意力集中在 Petasis 反应上 [7]。此前,我们已成功利用该反应及相关反应在螺环吡咯烷的合成中构建季碳中心 [8]。对于环化反应,我们选择了关环复分解反应。自 Morton 的开创性研究以来,关环复分解(RCM)方法是基于环化反应的 DOS 研究中最常用的方法之一 [9,10]。因此,在本研究中,我们报道了结合 Petasis 反应和 Grubbs 关环复分解反应,使用多种环酮作为起始原料,可扩展地合成药物化学相关螺环砌块的研究成果(图 1)。
2. 结果与讨论
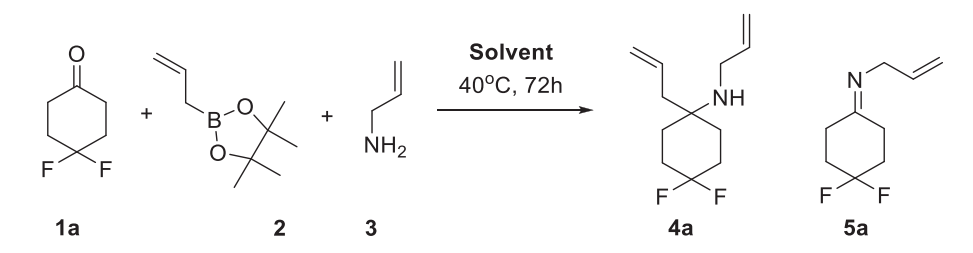
基于我们在合成 α,α- 二取代高烯丙基胺方面的经验,我们重点研究了 Petasis 反应、Hosomi–Sakurai反应以及格氏试剂与亚胺的加成反应序列,以在构建/偶联步骤中创建季碳中心。选择 Petasis 反应是因为它可以在一步反应中轻松引入两个烯烃片段 [11]。因此,我们开始以烯丙基硼酸频哪醇酯 2、烯丙胺 3 和 4,4 - 二氟环己酮 1a 为模型底物,对百克级规模的 Petasis 反应进行优化(方案 1)。选择上述酮 1a 是因为可以通过19F NMR 光谱轻松控制反应。起始酮 1a 和中间体亚胺 5a 在光谱中呈单峰,而产物 4a 为双取代 AB 体系,这使得反应混合物的定量分析变得简单(见支持信息)。筛选结果总结于表 1(优化过程见实验部分)。优化的条件是在甲苯中于 40°C 加热 72 小时,这使得目标产物 4a 几乎定量生成。所用试剂的比例为 1a:2:3=1:1.25:1.1(表 1,条目 10)。除三氯甲烷外,所有溶剂均可用于该反应。然而,在甲苯中的反应混合物更便于产物的分离和纯化。通过简单的水洗即可从粗产物中除去所有副产物和剩余的起始原料(硼酸、频哪醇等)。即使在 40°C 下,1M 的起始酮浓度在 72 小时内也足以达到 99% 的转化率。对于热稳定性较好的底物,可以加热到更高的温度(50-80°C),具体数值因化合物而异(详见实验部分)。对于这类衍生物,更高的温度可在更大规模下提供更高的产率。然而,对于不稳定的底物,如含氧杂环丁烷的 1f,40°C 的加热温度至关重要。
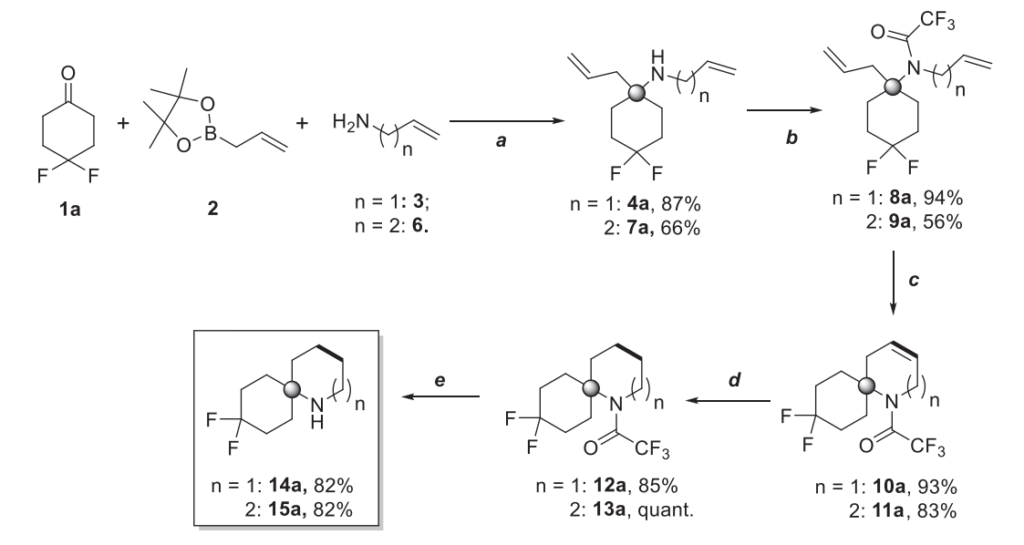
掌握了优化的 Petasis 反应方案后,我们研究了项目的下一步偶联和后修饰步骤,以得到药物化学相关的砌块。除了烯丙胺 3,高烯丙胺 6 也能以 66% 的制备产率得到 Petasis 产物 7a。得到的两种 α,α- 二取代底物 4a 和 7a 均用三氟乙酸酐(TFAA)保护,然后使用第二代 Grubbs 催化剂进行关环复分解反应。由于游离胺的碱性会使 Grubbs 催化剂中毒,氨基的保护基团性质至关重要 [12]。在形成 6 元和 7 元环的情况下,关环复分解反应在多克级规模上均能获得优异的制备产率。化合物 10a 和 11a 经进一步催化氢化,随后三氟乙酸脱保护,得到目标胺 14a 和 15a(方案 2)。
为了在氨基组分中引入官能团,由于相应的额外功能化不饱和胺的可获得性和稳定性不足,我们对策略进行了修改。在这种情况下,我们在 Petasis 步骤中使用苄胺,得到化合物 16。随后,后者与 2-(溴甲基)丙烯酸甲酯 17 进行烷基化反应,得到的产物 18 是用于关环复分解环化的优良起始功能化底物。作为起始的多样化酮库,我们选择了一组具有 C2v 对称性的模型底物,以避免在项目过程中形成非对映异构体混合物。其中包括最简单的丙酮 1b、C4-C6 环酮 1c-1e、氧杂环丁烷 1f、四氢 - 4H - 吡喃 - 4 - 酮 1g 和 Boc 保护的氮杂环丁烷 1h。一方面,这样的底物库为最终砌块带来了骨架多样性;另一方面,也使我们能够评估酮在拟议的 DOS 研究中的适用范围和局限性。
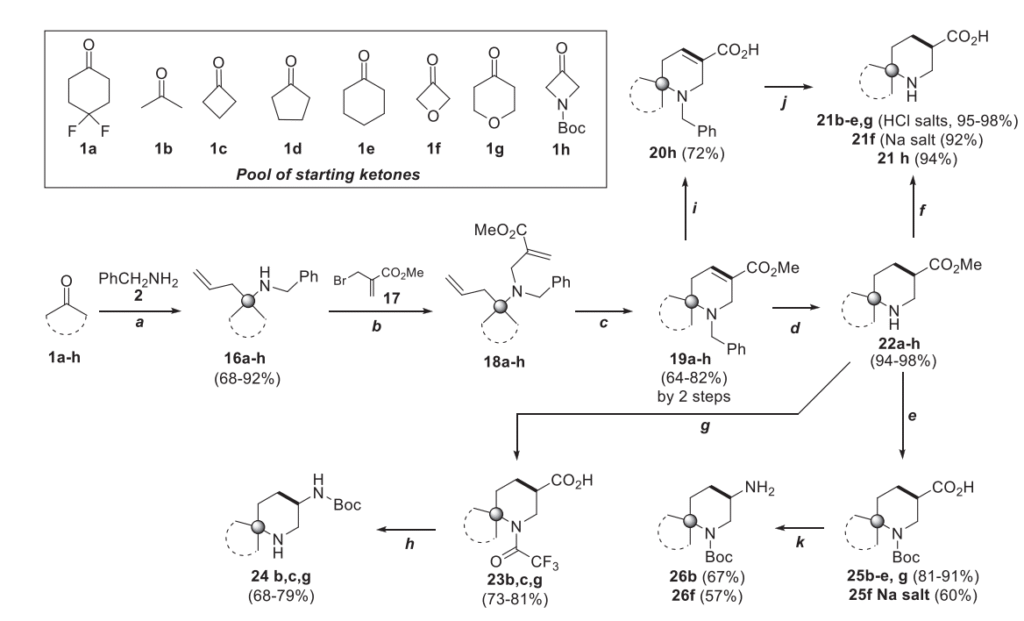
我们使用氟代模型酮 1a 验证了构建 / 偶联 / 配对序列。在烯丙基硼酸频哪醇酯 2 和苄胺的情况下,Petasis 步骤得到化合物 16a,分离产率为 74%。随后,16a 与溴化物 17 在无水乙腈中,以 K2CO3 为碱,在 80°C 下进行烷基化反应,得到化合物 18a。通过用甲基叔丁基醚稀释反应混合物、过滤生成的 KBr、蒸发有机相以及对残余物进行色谱纯化,得到化合物 18a(见支持信息)。该序列以 81% 的产率得到纯化合物 18a,将其在二氯甲烷中进行关环复分解反应,以 89% 的分离产率得到关键的双环产物 19a。与之前的底物 8a 和 9a 不同,化合物 19a 含有胺中心,这会使 Grubbs 催化剂中毒 [12]。考虑到在碳-碳双键存在下苄基的脱保护难度,我们通过加入等摩尔量的对甲苯磺酸一水合物将胺转化为盐,从而
“中和” 氮孤对电子的影响。我们还观察到色谱纯化并非必需,粗品 18a 也能以相当的制备产率得到目标产物 19a。因此,我们将其他类型的18化合物作为粗品用于环化步骤,无需额外纯化。在所有情况下,关键的双环化合物19b-h在多克级规模上均能以优异的制备产率得到(见支持信息和表 2,步骤 B + C)。氨基的脱保护通过在甲醇中于 1atm 氢气下用 10% Pd/C 氢化实现。在此过程中,碳 - 碳双键也会发生氢化,得到化合物 22a-h,产率几乎定量(表 2,步骤 D)。通过在 2M 盐酸水溶液中进行酸性水解,得到 “游离” 氨基酸 21b-e,g 的盐酸盐(表 2,步骤 F)。例外情况是含有对酸不稳定的氧杂环丁烷和 Boc - 氮杂环丁烷环的化合物 21f 和 21h。对于氧杂环丁烷衍生物,22f 的水解通过在无水甲醇中用 NaOH 实现,得到化合物 21f 的钠盐。在制备氮杂环丁烷 21h 的过程中,我们采用了稍复杂的序列:先用 NaOH 将酯 19h 水解为化合物 20h,产率 72%,然后进一步氢化得到目标产物 21h,产率 94%(方案 3)。化合物 22 的进一步后修饰包括常见的官能团转化,得到正交 Boc 保护的二胺 24 和 26,它们也是有价值的药物化学砌块 [13]。在这两种情况下,均使用 Curtius 重排将酸官能团转化为氨基(方案 3)。
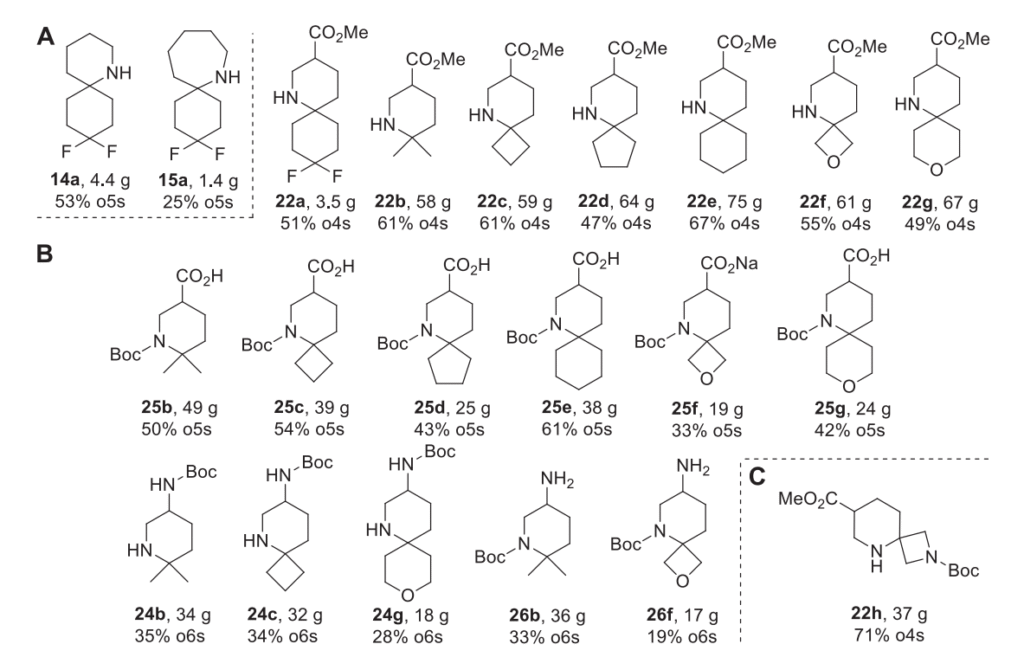
在该项目的合成部分,我们得到了一组包含 2 种单功能砌块 14a 和 15a(图 2,框 A)、18 种双功能砌块 22、24-26(图 2,框 B)和 1 种三功能砌块 22h(图 2,框 C)。所得化合物基于 12 种 Bemis-Murco 骨架。它们适用于 REAL 数据库产品 [14],并符合 Ro2 的要求 [15]。
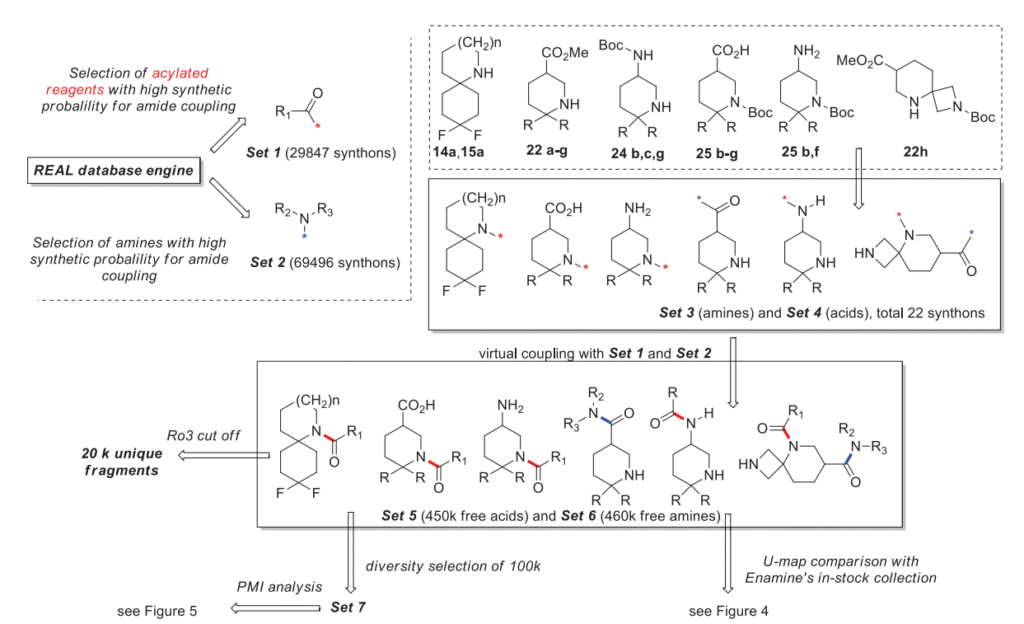
为了评估合成的砌块的重要性,我们进行了针对有形化学空间的计算机化学信息学研究,这些砌块通过 REAL 数据库引擎实现 [16]。首先,在这项模型计算机研究中,我们决定仅使用最流行的药物化学反应——酰胺偶联 [17]。但是,考虑到所获得的砌块具有正交保护的多官能性,为了通过虚拟偶联创建新的化学空间,我们使用了我们之前开发的用于高通量合成筛选化合物的一锅法连续偶联-脱保护-偶联方法 [15]。作为用于修饰本研究中设计的骨架的 “封端” 单功能砌块,我们选择了一组酸(组 1)和胺(组 2),根据我们之前的实验统计,它们在酰胺偶联中具有优异的化学可行性。所有这些酸和胺都被转化为相应的虚拟偶联合成子。然后从获得的砌块中生成一组合适的合成子:对于胺 14a 和 15a,对应于一步酰胺偶联;对于氨基酯 22,对应于与组 1 中脱保护的酯进行一锅两步酰胺偶联;对于二胺 24 和 26,对应于与组 1 中 Boc 脱保护的胺进行两步酰胺偶联;对于氨基酸 25,对应于与组 2 中 Boc 脱保护的胺进行一锅两步酰胺偶联;最后,对于二氨基酯 22h,对应于与组 1 中脱保护的酯进行一锅四步酰胺偶联,随后与组 2 中 Boc 脱保护的胺进行酰胺偶联。研究的下一步是使用制备的组对砌块进行枚举。然后,对所得化合物进行脱溶剂化和中和处理。这一步骤对于消除因盐和盐酸盐的存在而产生的所有重复项是必要的。结果,得到了一组约 45 万个独特化合物,其中原始砌块用作胺(组 5),另一组约 46 万个独特化合物,其中原始砌块用作酸(组 6)(图 3)。值得注意的是,所有生成的化合物都具有游离的官能团(胺或酸),可用于后续与组 1 和组 2 的迭代偶联。这使我们能够仅使用酰胺偶联反应就生成约 360 亿的化学空间(组 6× 组 1 + 组 5× 组 2 - 重复项)。如此庞大的空间分析需要大量的计算资源。同时,结构的新颖性可估计为该空间中的约 91 万个分子。
为了测试所获得的化学空间的物理化学性质,根据 “三规则” 对所得化合物组的理化参数进行了分析:分子量≤300、氢键受体≤3、氢键供体≤3、计算 logP≤3 [18]。该分析得到组 5 有 4052 个独特片段,组 6 有 8419 个独特片段,总共 12471 个独特片段。理化参数计算使用免费软件 DataWarrior 进行 [19]。
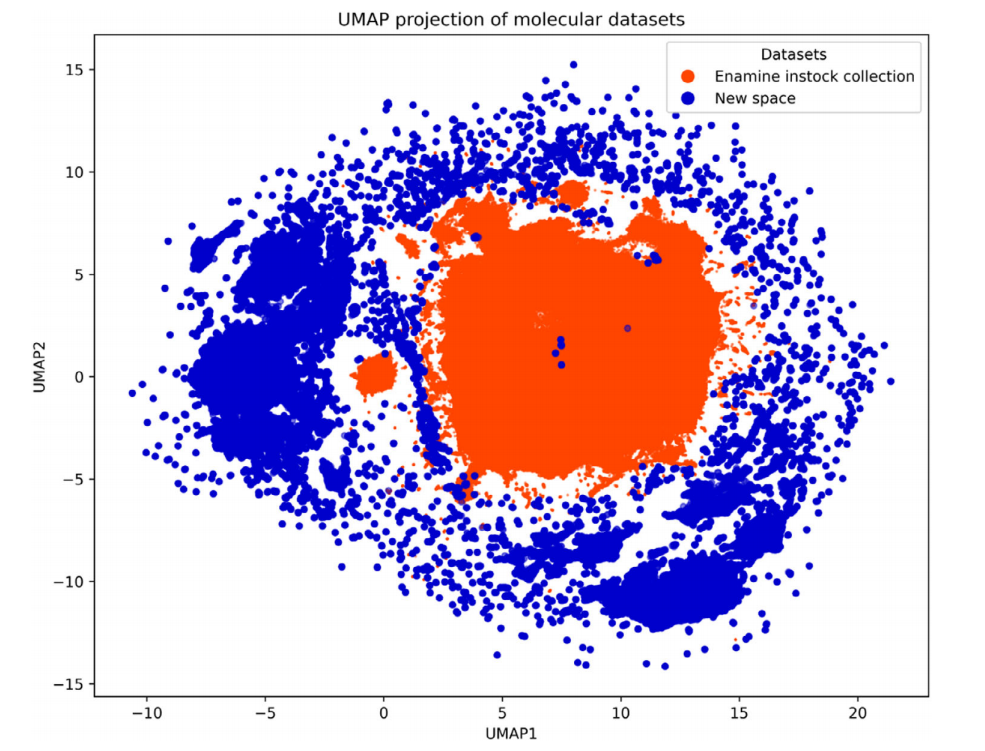
为了测试生成的有形化合物的新颖性,合并组 5 和组 6,使用均匀流形近似和投影(UMAP)方法进行分析,该方法广泛应用于数据可视化中的降维 [20]。这种降维算法使用分子特征作为输入数据生成 Rn 空间,然后对其进行降维,得到反映分子相似性的二维投影坐标(UMAP1 和 UMAP2)。这种方法通过将相似分子沿相应的 UMAP 坐标放置得更近,实现数据可视化。如图 4 所示,Enamine 库存筛选集合(橙色数据点)与我们枚举的化合物(蓝色数据点)的化学空间重叠极小,进一步证实了新砌块空间的独特性,这有助于从多个方向拓展化学空间。
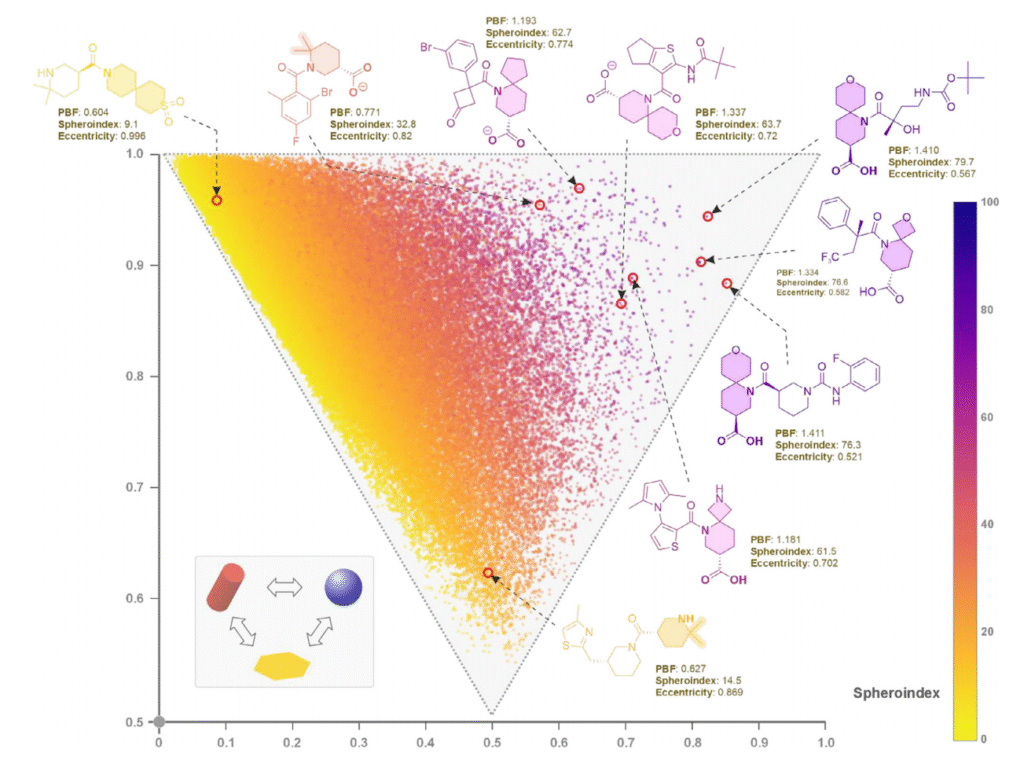
为了分析所得化合物形状的新颖性,考虑到该方法对计算能力的要求较高,我们使用 RDKit 库中的 MaxMinPicker 函数从整个库(组 5 和组 6)中选取了 10 万个多样化化合物 [21]。然后,使用 RDKit 为每个化合物生成多达 50 个构象,从中选择能量最低的构象进行进一步分析。如图 5 所示,使用这些砌块能够生成具有前景形状的化合物,如球形和盘形。这一结果非常有前景,因为之前对 ZINC 数据库的分析表明,大多数(约 75%)“常规” 化合物具有线性(1D)或平面(2D)形状 [22]。
3. 结论
最后,我们开发了一种基于 Petasis/Grubbs 反应序列的多克级规模 3D 形状砌块的类 DOS 合成方法。生成季碳中心的 Petasis 反应经优化后获得了优异的制备产率。后续优化的关环复分解反应能够以制备规模形成螺环分子骨架。环酮与功能化烯丙胺和高烯丙胺的不同组合为基于 16 种 Bemis-Murco 骨架的功能化螺环哌啶和氮杂䓬的合成开辟了道路。对这些砌块在药物化学相关酰胺偶联中的应用进行的计算机化学信息学研究表明,所获得的有形化合物在化学空间的广泛探索中具有独特性。这项研究清楚地表明,在砌块层面使用类 DOS 方法,并结合成熟的有形阵列方法(如 Enamine 的 REAL 数据库),在利用超大有形化学空间加速未来药物发现方面具有巨大潜力 [22]。