
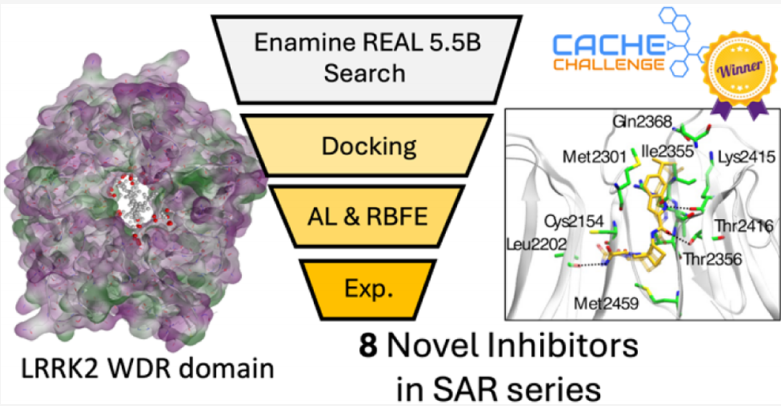
编者按:上期我们介绍了一篇利用 Enamine 实体化合物数据开展虚拟筛选的文章,本期将为大家呈现一篇采用 Enamine 虚拟数据库 Real Space 进行高通量虚拟筛选的研究成果。该文作者所在的课题组在 CACHE 挑战赛 #1 中斩获最高分,本文正是对这一获奖工作的相关总结与详细介绍。研究中,团队对包含约 55 亿个化合物的 Enamine 虚拟数据库 Real Space 进行高通量虚拟筛选,最终成功发现 8 个能与 LRRK2 WDR 结构域结合的先导化合物。
摘要
富亮氨酸重复激酶 2(LRRK2)是家族性帕金森病中突变率最高的基因,其突变会导致该疾病的 pathogenic 特征。LRRK2 的 WDR 结构域是一个尚未被充分研究的帕金森病药物靶点,在计算命中发现实验关键评估(CACHE)挑战赛的第一阶段之前,尚无已知的抑制剂。CACHE 挑战赛的一个独特优势是,预测的分子会在内部进行实验验证。在此,我们报告了 LRRK2 WDR 抑制剂分子的设计和实验确认。我们采用了基于主动学习(AL)的机器学习(ML)工作流程,该流程基于优化的自由能分子动力学(MD)模拟,并利用热力学积分(TI)框架,围绕我们之前确认的两个命中分子扩展化学系列。在 35 个经过实验测试的分子中,我们确定了 8 个经过实验验证的新型抑制剂(命中率为 23%)。这些结果证明了我们基于自由能的主动学习工作流程能够快速高效地探索大型化学空间,同时最大限度地减少昂贵模拟的数量和时长。该工作流程广泛适用于筛选任何化学空间中的高亲和力小分子类似物,但需遵循相对结合自由能计算的一般约束。TI MD 计算的平均绝对误差为 2.69 kcal/mol,相对于命中化合物的实测解离常数(KD)而言。
引言
计算命中发现实验关键评估(CACHE)挑战赛是一系列科学竞赛,旨在基准测试识别能够与特定分子靶点结合的小分子的计算方法,其目标是推动针对罕见和未被充分研究的重要药用靶点的计算机药物发现工作1。CACHE 挑战赛 #1 的目标是寻找与富亮氨酸重复激酶 2(LRRK2)的 WDR 结构域结合的小分子结合剂,LRRK2 是一种多结构域蛋白,也是帕金森病(PD)的靶点。LRRK2 是家族性 PD 中突变最频繁的蛋白质,其突变会导致该疾病的 pathogenic 特征2-4。虽然已有针对 LRRK2 激酶结构域的抑制剂和 PROTACs 的相关报道5-7,但迄今为止,尚无配体靶向该蛋白的相邻 WDR 结构域8,尽管 WDR 结构域已被证明在其他蛋白质中具有成药性⁹。一个反复出现的致病性突变位于 LRRK2 WDR 二聚体的界面,突显了该结构域与疾病的相关性8。CACHE 挑战赛 #1 包括两轮预测和实验确认,允许参与者将第一轮获得的见解纳入第二轮预测中。第一阶段侧重于通过计算机筛选市售文库进行命中识别10-11,随后进行实验测试,从我们的选择中产生了 5 个已确认的命中分子12。
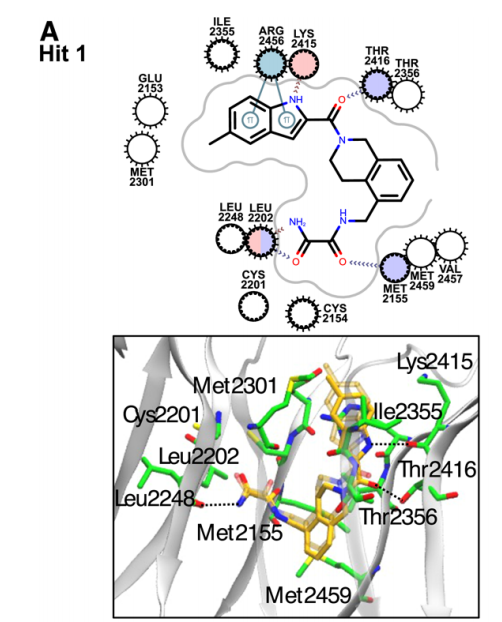
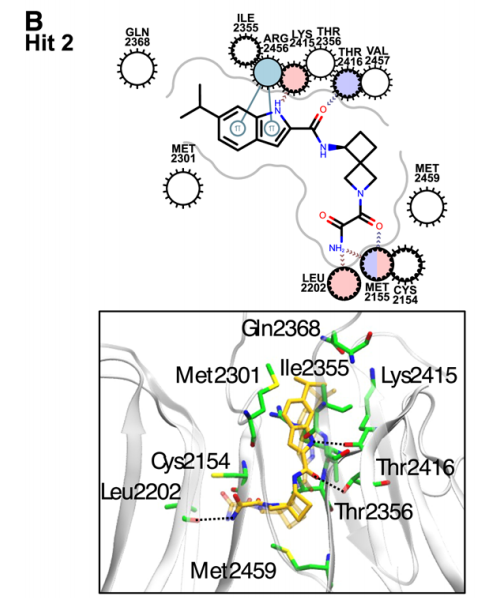
在此,我们报告了关于两种 LRRK2 WDR 结构域结合剂的优化研究的获奖成果。这些配体,即命中分子 1 和命中分子 2(图 1),在 “第一轮” 中最初验证的 5 个命中分子中具有最高的实验结合亲和力。计算流程包括从与我们的命中分子具有共同亚结构的市售文库中进行选择,然后进行对接和优化的分子动力学(MD)热力学积分(TI)模拟13,并在主动学习(AL)14的指导下计算化合物与靶点的相对结合自由能。我们筛选了约 55 亿种市售化合物(Enamine Database, 截至 2022 年 10 月发布),选择了约 25,000 种配体用于 AL-RBFE 计算,并计算了 672 种配体的相对结合自由能。基于计算的相对结合自由能,选择了 75 个分子进行湿实验验证,其中 35 个进行了进一步的实验测试。通过表面等离子体共振(SPR)和氟代分子的19F 核磁共振(NMR)确认了 8 个配体与 WDR 结构域的结合。
这些发现为下一步发现靶向 LRRK2 的 WDR40 结构域的高亲和力选择性化合物铺平了道路。我们的化合物选择获得了由生物物理学和药物化学行业专家组成的独立委员会在 CACHE 挑战赛 #1 竞赛中的最高分15。
结果与讨论
计算流程
我们开发了一个全面的命中优化流程,利用我们的主动学习(AL)工作流程进行相对结合自由能(RBFE)计算14(参见方法部分 4)。Enamine REAL 数据库10用于虚拟筛选,在本工作开始时该数据库包含 55 亿个小分子化合物。虚拟筛选协议的第一步是使用每个命中分子的两种不同 SMARTS 模式过滤该集合(图 2B):第一种模式包含 Murcko 骨架和草酰胺基团,第二种模式仅包含源自初始命中分子的 Murcko 骨架。这产生了两组分子:i)一组最接近的类似物和 ii)一组更远的类似物,以下称为 “一般类似物”。对于最接近的类似物(250 个分子),我们对与命中分子 1 和命中分子 2 相关的蛋白质-配体复合物的 MD 代表性结构进行了模板对接。因此,选择了 214 个对接分子(46 个命中分子 1 的类似物和 168 个命中分子 2 的类似物)用于相对结合自由能 MD 模拟。为了提高命中分子 1 的最接近类似物的多样性,我们在一般类似物中进一步进行最近邻(NN)搜索,以识别与计算出的相对结合自由能最低的前 9 个分子具有高度相似性的分子。这 27 个已识别的分子随后被纳入最接近的类似物集合中(详见第 4.1.1.2 节),并以与初始批次类似的方式进行对接和相对结合自由能计算。相对结合自由能被转换为绝对结合自由能(详见方法部分 4)。该集合以下称为 AL 前集合。
对于一般类似物(约 340,000 个分子),我们首先在没有模板的情况下进行对接,并通过对接分数进行过滤。随后,我们使用与最接近的类似物相同的协议进行带模板的对接。接下来,我们基于对接分数和相对于模板的均方根偏差对对接的配体进行额外过滤。所得集合包含约 16,000 个命中分子 1 的类似物和约 9,000 个命中分子 2 的类似物。该集合(约 25,000 个分子)被称为 AL 集合(见图 2)。
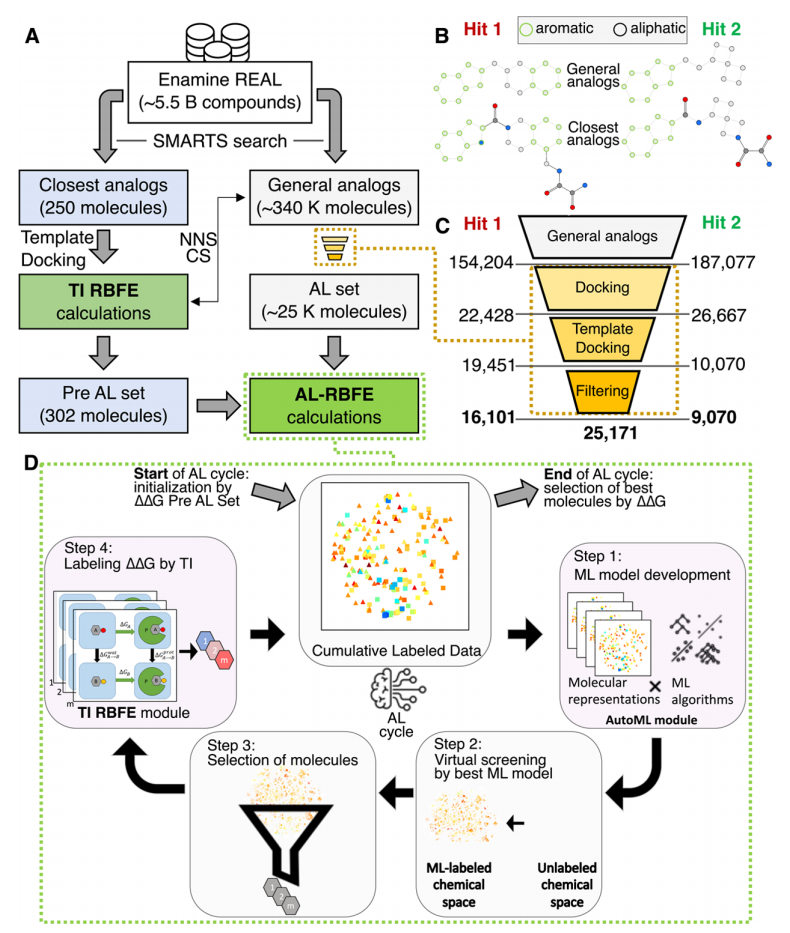
(详见正文描述)。对应于最近相似物、通用相似物和RBFE计算的模块分别以蓝色、灰色和绿色显示。NNS表示最近邻搜索,S表示人工筛选(详见方法部分4)。(B) 用于Hit 1和Hit 2的最近相似物和一般相似物的SMARTS模式。 (C) Hit 1和Hit 2一般相似物的虚拟筛选。管道每个步骤后Hit 1和Hit 2的分子数量如图所示。(D) 由AL引导的RBFE计算的自动化计算工作流的总体方案(AL-RBFE)。工作流包括两个主要模块:AutoML和MD TI RBFE,以及四个主要步骤。化学空间以2D t-SNE图显示。Hit 1和Hit 2的类似物及其计算的ΔΔG值以彩色正方形和三角形表示,与图4中的颜色方案一致。
主动学习的理论背景以及 AL-RBFE 工作流程的详细概述在我们之前的工作中提供 14。简而言之,AL-RBFE 工作流程是一个迭代过程,在每次迭代中,使用具有计算出的 MD TI 相对结合自由能的分子来训练机器学习模型,该模型基于其化学结构预测配体的相对结合自由能。机器学习模型训练完成后,它预测数据集中所有分子的相对结合自由能,并使用这些预测来选择用于下一轮 MD TI 模拟的相对结合自由能计算的分子。在这项工作中,我们使用绝对结合自由能而非相对结合自由能,以便使用相同的机器学习模型筛选两种命中分子的类似物。重要的是,我们仍然进行 MD TI 模拟来计算相对结合自由能,而不是绝对结合自由能,之后我们将相对结合自由能转换为绝对结合自由能(详见方法部分 4),然后使用这些数据训练机器学习模型。
2.2 相对结合自由能计算的扰动图
图 S1 显示了命中分子 1 类似物的 MD TI 相对结合自由能计算的扰动图。对于 AL 前集合中的所有命中分子 1 类似物,使用命中分子 1 作为参考配体进行相对结合自由能计算。相比之下,使用配体 X 作为参考配体进行一般类似物的相对结合自由能计算。配体 X 源自命中分子 1 的类似物 A,其在 AL 前集合的分子中具有最低的绝对结合自由能(见图 S1)。由于难以准备相对结合自由能计算的初始结构,配体 X 被用作一般类似物的参考配体。具体而言,当试图计算 AL-1 集合中某些分子的相对结合自由能时,我们发现这些取代基在 MD TI 输入结构中的初始构象严重扭曲。这种扭曲的原因是,在准备 MD TI 相对结合自由能模拟的系统时,1,2,3,4-四氢异喹啉的 5 位取代基被迫与参考配体(命中分子 1)的甲基草酰胺基团对齐(详见方法部分 4)。这种人工对齐可能会偏向该配体的采样,并提供不可靠的相对结合自由能。
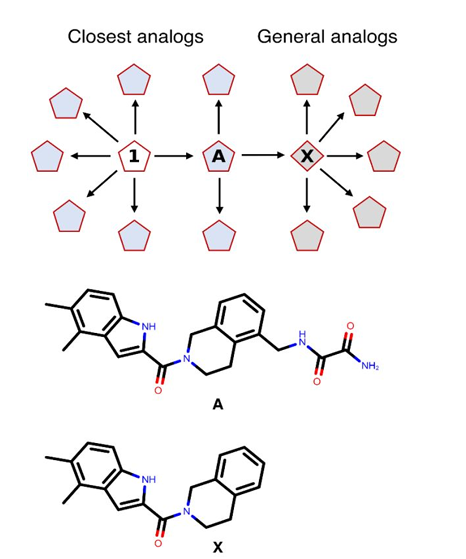
S1. 用于计算Hit 1相对结合自由能的扰动图。Hit 1以白色五边形表示。提供了Hit 1最接近的类似物A和中间体化合物X的结构。
此外,在某些情况下,这会导致配体与蛋白质之间出现冲突,随后 MD 模拟失败。为了应对这一挑战,我们通过将甲基草酰胺基团替换为氢,从配体 A 创建了配体 X。使用配体 X 作为相对结合自由能计算的参考,使具有挑战性的配体的取代基能够保持其初始构象。同时,它还允许对在 1,2,3,4-四氢异喹啉的 5 位没有取代基的配体进行更优化的相对结合自由能计算。虽然在这些情况下使用命中分子 1 作为参考配体需要完全消除甲基草酰胺基团,但使用配体 X 作为参考可以避免这一过程,从而提高计算的收敛性。
2.3 主动学习引导的相对结合自由能计算结果
我们执行了八次 AL-RBFE 工作流程迭代。AL 前集合被用作训练集,在第一次迭代中构建初始配体绝对结合自由能预测机器学习模型。对于接下来的七次 AL 迭代(AL-1−AL-7),通过机器学习模型选择用于相对结合自由能计算的分子(每个 AL 迭代选择的化合物数量详见表 S2)。由于命中分子 1 比命中分子 2 具有更高的结合亲和力,AL-1−AL-6 迭代仅针对命中分子 1 的类似物进行。最后一次迭代 AL-7 包括命中分子 1 和命中分子 2 的类似物,旨在通过命中分子 2 的类似物丰富预测的命中分子。每轮 MD TI 模拟后,计算出的相对结合自由能被转换为绝对结合自由能(详见方法部分 4)。
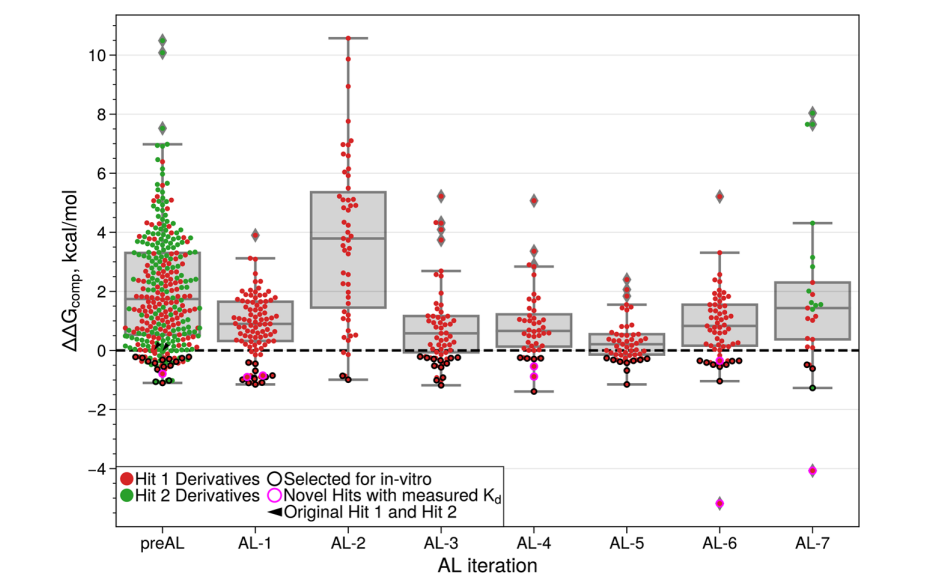
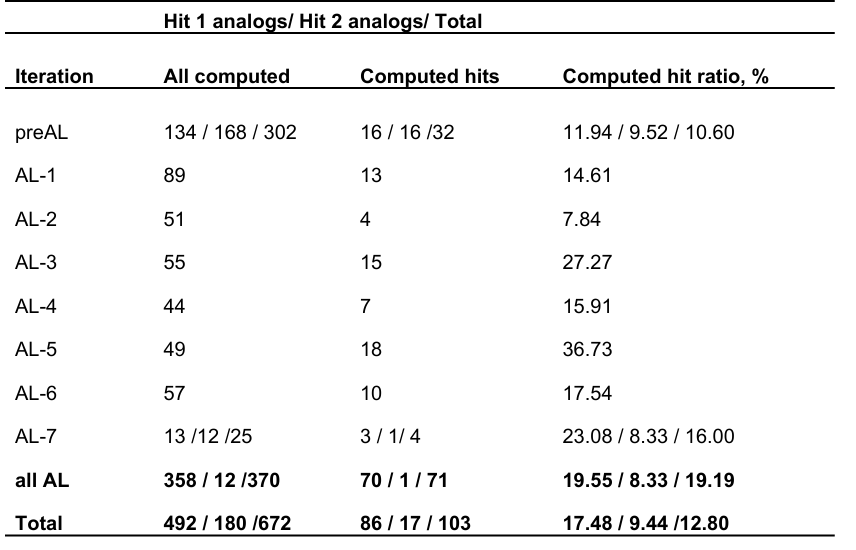
AL-RBFE 工作流程所有迭代的结果如图 3 和表 S2 所示。总共计算了 674 个分子的绝对结合自由能(分别为 493 个和 181 个命中分子 1 和命中分子 2 的类似物)。总体而言,我们确定了 102 个计算出的绝对结合自由能低于初始命中分子的类似物(分别为 87 个和 15 个命中分子 1 和命中分子 2 的类似物)。对于命中分子 1,约 80% 的具有改进绝对结合自由能的类似物是由 AL 选择的(87 个分子中的 70 个),其余则来自 AL 前集合。在每次 AL 迭代中都发现了改进的类似物。在所有计算的命中分子 1 类似物中,AL 集合中具有改进绝对结合自由能的命中分子 1 类似物的比例是 AL 前集合的 1.5 倍以上:分别约为 20% 和 13%。这些结果证明了利用 AL 指导相对结合自由能计算的有效性。
为了更深入地了解 AL-RBFE 工作流程的性能,我们使用 t-SNE 投影可视化了所有类似物(AL 前和 AL 集合一起)和具有计算出的绝对结合自由能的分子的化学空间(图 4)。为每个单独的 AL 迭代(图 4A)以及所有迭代的结果(图 4B)构建了 t-SNE 图。我们可以看到,AL 前集合的计算分子分布在化学空间的不同区域,而不是集中在同一区域,这表明该集合的分子之间存在一定的结构多样性。在其余迭代过程中保持相同的趋势:AL 从化学空间的不同区域选择分子,包括先前迭代筛选的区域和先前阶段未探索的区域。因此,使用 AL 指导分子选择增加了具有改进绝对结合自由能的配体的多样性。
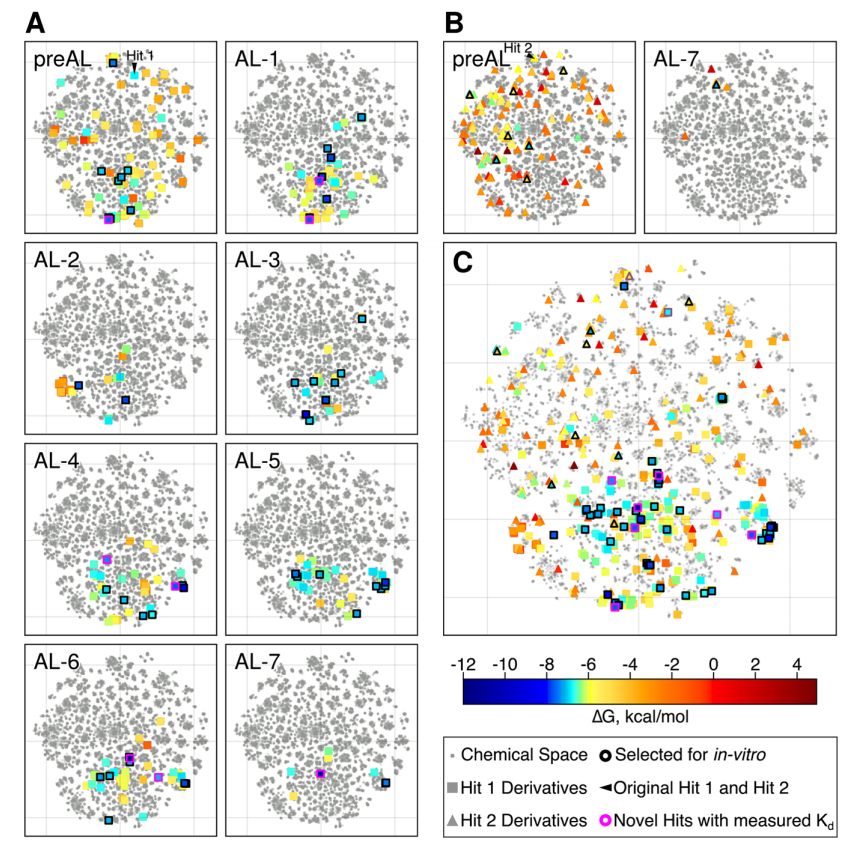
2.4 实验验证的命中分子
根据挑战预算(75 个分子或 10,000 美元,以先到者为准)选择提交用于实验测定的分子,具体如下:70 个分子仅基于计算出的最负的 ΔG 贪婪选择(67 个命中分子 1 的衍生物和 3 个命中分子 2 的衍生物),其余 5 个分子在具有负 ΔG 但偏向化学多样性的命中分子 2 的衍生物中选择。选定的 75 个分子向 Enamine 化学供应商询价,其中 35 个被采购并通过表面等离子体共振(SPR)在 50μM 下进行实验测试(详见方法部分 4、实验方法部分 4.5 和表面等离子体共振部分 4.5.2)。所有 Enamine 提供的化合物通过 HPLC 分析纯度均 > 95%。11 个命中候选物进入剂量反应实验,其中 8 个具有可测量的解离常数 KD 优于 250μM,并且具有可接受的 SPR 传感器图(Chi²<10% Rmax;T (KD)>1),KD 值范围为 18 至 23μ0M(图 5 和表 S1)。由于一些命中分子是氟代分子,我们使用 19F-NMR 作为正交测定法来确认结合不是测定特异性的(图 5)。我们在动态光散射测定中验证了化合物在相关浓度下可溶且不聚集(图 5 和表 S1)16。因此,CACHE 挑战赛第二轮的命中率为 23%。CACHE 通常会丢弃结合率低于 30% 的 SPR 命中分子。尽管化合物 O1 显示出 27% 的结合率,低于该 cutoff,且化合物 O2 未显示出 19F-NMR 的结合信号,但它们属于已确认与 LRRK2 结合的化学系列,并具有可接受的质量描述符(Chi²<10% Rmax 和 T (KD)>1)。因此,它们被纳入报告的构效关系(SAR)中(图 5)。
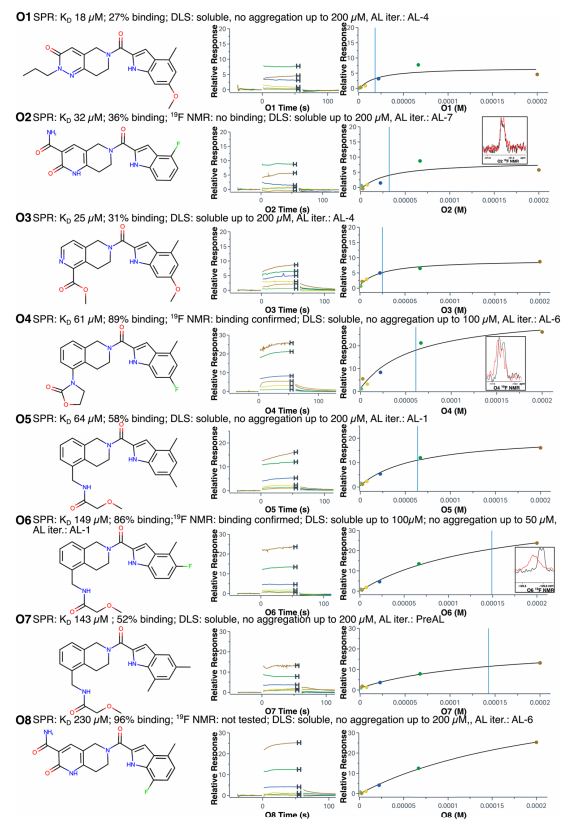
O1−O8 命中分子的结合自由能如表 S3 所示。对于两个命中分子(O4、O6),通过 19F - 核磁共振(NMR)进一步确认了结合。实验解离常数(表 S3 和表 S1)范围为 18μM(尽管该化合物通过 SPR 仅显示 27% 的结合)至 230μM(相应的绝对结合自由能范围为 - 5.0 至 - 6.5kcal/mol),其中三个更强的结合剂(KD=18-32μM),两个中等结合剂(KD=61-64M)和四个弱结合剂(KD=143-230μM)。命中化合物的计算绝对结合自由能与实验绝对结合自由能之间的平均绝对误差为 2.69kcal/mol。这证明了我们提出的工作流程能够识别和扩展具有可靠构效关系(SAR)的系列,即使在本工作进行时没有实验确认的结合构象的新型靶点上也是如此。在八个结合剂中,只有一个是从 AL 前集合中识别出来的,尽管该集合包含近一半的模拟化合物,这表明主动学习在选择结合剂方面比 naive 选择方案更有效;然而,由于涉及的样本量较小,我们无法就两种选择方法得出具有统计显著性的结论。
所有优化的命中分子都是命中分子 1 的类似物,包括通过羰基桥与哌啶环连接的吲哚环。四个命中分子(O4、O5、O6 和 O7)还包含与哌啶环连接的苯环,形成 1,2,3,4-四氢异喹啉系统。这五个命中分子中的三个(O5、O6 和 O7)具有相同的修饰(草酰胺基团的末端酰胺被甲氧基甲基取代),仅在吲哚环中的取代基不同。命中分子 O7 来自 AL 前集合,另外两个命中分子(O6 和 O5)在第一次迭代(AL-1)中被 AL 选择。计算出的绝对结合自由能和实验绝对结合自由能均在 0.6kcal/mol 以内,其中命中分子 O4 在这些命中分子中显示出最强的结合亲和力。化合物 O4 与其他命中分子不同:甲基草酰胺被恶唑烷酮环取代。这是唯一包含三环系统的优化命中分子。尽管它具有最高的计算绝对结合自由能,但它显示出中等的实验结合亲和力,KD 为 61μM。
在其他四个化合物(O1、O2、O3、O8)中,1,2,3,4-四氢异喹啉系统的取代苯环被取代的六元杂环芳香环取代。命中分子 O8 和 O2 包含 2-吡啶酮甲酰胺,化合物 O1 包含 N-丙基哒嗪-3-酮,命中分子 O3 包含被甲基羧酸盐基团取代的吡啶。化合物 O8 和 O2 在 AL 工作流程的最后两次迭代中被选择,并且与其余类似物相比具有显著更低的绝对结合自由能。这些类似物仅在吲哚环上的取代基不同;然而,虽然命中分子 O2 在所有命中分子中显示出第三强的结合亲和力,KD 为 32μM,但命中分子 O8 是相当弱的结合剂,KD 为 230M。值得注意的是,我们的 MAE 值受这两种化合物的严重影响。排除这些化合物后,MAE 为 1.66kcal/mol。
在第四次 AL 迭代(AL4 集合)中选择的命中分子 O1 和 O3 在吲哚环中具有相同的取代基,但在取代的环系统(异丙基哒嗪-3-酮与甲基吡啶羧酸盐)中不同。尽管存在结构差异,但这些命中分子的计算绝对结合自由能和实验绝对结合自由能彼此相对接近(差异小于 0.4kcal/mol)。命中分子 O1 是所有命中分子中最强的结合剂,KD 为 18M,命中分子 O3 是第二强的结合剂,KD 为 25μM。重要的是,所有三个最强的结合剂(O1、O2、O3)都属于命中分子 1 的一般类似物集合,并且具有相对多样的结构。
3. 结论
分子建模和机器学习技术的最新进展为开发生物医学应用中的新型计算方法铺平了道路。本文进一步证明了 AL-RBFE 组合方法14在识别和扩展小分子结合剂系列至靶蛋白方面的实用性。该方法结合了精确的基于物理的分子模拟和机器学习方法,即使对于具有有限先前信息的具有挑战性的靶点也是有效的。
CACHE 是一项公私合作计划,旨在评估和改进用于识别具有药物重要性的分子靶点的小分子结合剂的计算方法1。迄今为止,已组织了六次 CACHE 挑战赛17。每次 CACHE 挑战赛包括两轮预测,因此允许参与者利用从初始轮次获得的见解进行后续设计工作。在 CACHE 挑战赛 #1 中,参与者被要求预测与 LRRK2 的 WDR 结构域的中央腔结合的小分子。在挑战赛开始时,尚无已知的与 LRRK2 WDR 结构域结合的小分子化合物。参与者在第一阶段提交并通过结合测定实验确认的化合物被选作第二阶段优化的起点。在该阶段,要求参与者选择一组新的分子进行实验表征。
在这项工作中,我们开发了一个用于 SAR 系列扩展的计算流程,并将其应用于我们先前在 CACHE 挑战赛 #1 第二轮中识别的命中化合物。在挑战赛第一阶段实验确认的两个 LRRK2 WDR 结构域的小分子结合剂被选作计算机筛选市售类似物的母体分子。该流程整合了完善的计算方法,如化学亚结构搜索和分子对接,以及我们最近开发的用于 AL 指导的 MD TI 相对结合自由能计算的工作流程和我们的 TI 模拟时间优化算法。通过亚结构搜索以及随后对接和过滤约 55 亿个市售小分子化合物,我们获得了一组约 25K 个初始命中分子的类似物。利用具有优化模拟的 AL-RBFE 工作流程能够有效探索该集合,寻找具有改进的预测结合亲和力的类似物。我们通过仅计算 672 个类似物的 MD TI 相对结合自由能,识别出 102 个预测的命中分子。基于计算的 MD TI 相对结合自由能选择的一组 75 个预测命中分子被提交用于实验测试。在测试的 35 个分子中,结合测定揭示了 8 个命中化合物。虽然这些分子的结合亲和力仅在中等微摩尔范围内,但它们在正交测定中的确认结合、主要命中分子对无关靶点的选择性12-15,以及它们经实验验证的在高浓度下的溶解度和无聚集性,为进一步优化奠定了坚实的基础。
因此,我们在 CACHE 挑战赛 #1 第一轮和第二轮的结果表明,所提出的方法对于具有挑战性的蛋白质靶点的小分子结合剂的计算机设计是有效的。仅从已知的脱辅基蛋白结构开始,我们首先能够使用深度对接结合 MD TI 绝对结合自由能计算识别 5 个具有不同支架的结合剂12,然后使用我们的 AL-RBFE 工作流程显著扩展其中一个 SAR 系列。我们相信这种方法在简化和加速药物发现的早期阶段具有广阔的潜力。
4. 方法
4.1 数据库筛选和文库制备
计算流程包括两个集合的虚拟筛选:AL 前集合,包含命中分子 1 和 2 的最接近类似物;以及 AL 集合,包含命中分子 1 和 2 的一般类似物(见图 2A)。AL 前集合和 AL 集合均用于 AL-RBFE 计算。我们的流程的各个步骤描述如下。
4.1.1 最接近类似物的虚拟筛选
4.1.1.1 SMARTS 搜索阶段。在 Enamine REAL(截至 2022 年 10 月发布)数据库10中搜索命中分子 1 和 2 的最接近类似物,该数据库包含 55 亿个枚举化合物。使用 OpenEye 化学工具包通过命中分子 1 和 2 的亚结构的 SMARTS 模式(见图 2B,最接近的类似物)进行此搜索 18。SMARTS 模式基于命中分子的化学结构,但允许任何重原子取代,同时保留芳香性和药效团基团(草酰胺、肽键和芳香氮)。该搜索产生了 58 个和 192 个命中分子 1 和 2 的最接近类似物,分别。计算了这些文库中所有分子的 MD TI 相对结合自由能。
4.1.1.2 最近邻搜索(NNS)。在选择时,所有具有计算出的负相对结合自由能的命中分子 1 类似物均用作针对命中分子 1 一般类似物集合的查询分子(图 2A)。对于每个查询分子,从命中分子 1 类似物的模板对接后文库(n=19,451)(图 2C)中获取 3 个最近邻(基于 ECFP6-2048 位指纹上的 Tanimoto 距离),形成 27 个额外的独特分子列表用于相对结合自由能计算。
4.1.1.3 精选(CS)。在通过 SMARTS 搜索选择的初始分子集合的相对结合自由能计算之后,进行精选(CS,见图 2)。配体 A 是命中分子 1 的类似物,具有计算出的最低相对结合自由能(图 S1),用作母体化合物。基于命中分子 1 的一般类似物的目视检查和 MD TI 相对结合自由能,选择配体 A 的另一组类似物。所有选择的分子都具有 4,5-二甲基吲哚环,但在 1,2,3,4-四氢异喹啉环上的取代基不同。这产生了另外 49 个用于 MD TI 相对结合自由能计算的分子。
4.1.1.4 AL 前集合。AL 前集合(图 2)包括从 SMARTS 搜索以及 NNS 和 CS 阶段获得的所有命中分子 1 和 2 的最接近类似物,具有计算出的 MD TI 相对结合自由能。总体而言,AL 前集合包括 302 个分子:134 个命中分子 1 的类似物和 168 个命中分子 2 的类似物。
4.1.2 一般类似物的虚拟筛选
4.1.2.1 SMARTS 搜索阶段。在 Enamine REAL(截至 2022 年 10 月发布)数据库10中搜索命中分子 1 和 2 的一般类似物,该数据库包含约 55 亿个枚举化合物。使用 OpenEye 化学工具包通过命中分子 1 和 2 的亚结构的 SMARTS 模式(见图 2B,一般类似物)进行一般类似物搜索18。SMARTS 模式基于命中分子 1 和命中分子 2 的 Murcko 骨架,但允许任何重原子取代,同时保留芳香性模式。这形成了用于命中分子 1 和 2 的一般类似物的对接阶段的文库,分别为 154,204 和 187,077 个分子。
4.1.2.2 无模板对接阶段。使用 Glide SP 将选择的配体对接至 LRRK2 WDR 结构域的最小化晶体结构19(PDB ID:6DLO)。为每个分子保存三个具有最佳对接分数的对接构象。为对接准备的蛋白质结构和 Glide SP 的参数与 CACHE 挑战赛 #1 的第一阶段相同 ¹²。基于对接分数和对接构象的吲哚环样亚结构相对于相应命中分子的 MD 代表性构象的吲哚环的均方根偏差(RMSD indole),分别对命中分子 1 和 2 的衍生物的对接分子进行过滤(详见分子动力学部分 4.2.1)。过滤包括以下步骤:1)对于每个分子,选择具有相对于 MD 代表性构象的最小 RMSD indole 的最佳构象;2)保留满足 RMSD indole≤5Å 且 Glide 对接分数≤-6 的分子。因此,我们生成了用于进一步模板对接的文库,分别为 22,428 个和 26,667 个命中分子 1 和 2 的分子。
4.1.2.3 模板对接阶段。使用 OpenEye Make Receptor 程序(版本 4.0.0.0)准备与命中分子 1 和 2 复合的 LRRK2 WDR 结构域的 MD 代表性结构用于模板对接。命中分子 1 和命中分子 2 被设置为模板,并且不添加约束。使用 OpenEye OMEGA(版本 4.1.0.0)从 SMILES 生成 3D 构象。单个分子的最大构象数为 2000,最小均方根偏差(RMSD)为 0.2Å。使用 OpenEye HYBRID(版本 4.0.0.0)进行模板对接。对于每个分子,100 个最佳构象存储在输出数据中。所有其他参数均设为默认值。
基于对接分数和分子的广义 Murcko 骨架相对于命中分子 1 或 2 的 MD 代表性构象的相应亚结构的 RMSD(RMSD Murclo),对接的分子被过滤。过滤包括以下步骤:1)对于每个分子,选择一个具有最小 RMSD Murcko 的构象;2)保留满足 RMSD Murcko≤4Å、OpenEye Hybrid 对接分数≤-6 以及 OpenEye Hybrid 对接分数冲突分量≤0.5 的分子。这形成了分别为 19,451 个和 10,070 个命中分子 1 和 2 的衍生物的文库,总共 25,171 个分子。基于异构 SMILES 和带电分子,对文库进行额外的重复过滤。这形成了最终的文库,分别为 16,101 个和 9,070 个命中分子 1 和 2 的衍生物,总共 25,171 个分子。该文库将被称为 AL 集合。
4.2 Al 化学亲和力自由能计算。
4.2.1 分子动力学
在 CACHE 挑战赛 #1 阶段获得的与命中分子 1 和 2 复合的 LRRK2 WDR 结构域的对接结构被用作 MD 模拟的初始结构12。蛋白质-配体复合物在矩形水盒中溶剂化,盒边缘与溶质之间的最小距离为 12Å。蛋白质和水分别使用 FF14SB20力场和 TIP3P²¹ 模型进行参数化。配体原子参数使用 GAFF2²²(版本 2.11)获得,配体原子电荷使用 AM1-BCC23-24方法导出。使用 AMBER 20 的 pmemd.cuda 模块进行 GPU 加速的 MD 模拟25-27。模拟协议包括以下步骤:1)使用梯度下降法进行 2000 步最小化;2)在 NVT 系综中从 1K 加热至 298K,持续 100ps;3)在 NPT 系综中进行 300ps 的密度平衡;4)在 NVT 中进行 100ns 的生产模拟。在最小化和加热期间,对蛋白质、配体的重原子和位于结合位点的三个水分子施加谐波 RMSD 约束,并在密度平衡期间逐渐移除。在生产模拟期间不使用约束。生产 MD 模拟的前 10ns 被丢弃。通过平均最后 90ns 模拟的配体重原子和蛋白质 Cα 原子的坐标获得平均结构。选择配体重原子和蛋白质 Cα 原子相对于平均结构具有最小 RMSD 的轨迹帧作为代表性结构。
4.2.2 配体制备和参数化
参考分子和目标分子之间的原子映射基于它们相应的对接构象:通过 RDKit 获得最大共同亚结构,映射的重原子之间的最大距离为 1.1Å。使用 FESetup28v.1.2.1 软件包,以生成的原子映射和命中分子 1 和 2 的蛋白质 - 配体复合物的 MD 代表性结构作为输入数据,生成蛋白质 - 配体复合物和溶剂化配体系统的拓扑和输入坐标。使用 GAFF2 力场对配体进行参数化,通过 AM1-BCC 电荷模型分配电荷。使用 FF14SB 力场对蛋白质进行参数化,并采用 TIP3P 水模型。蛋白质 - 配体复合物和溶剂化配体盒大小与 CACHE 挑战赛 #1 阶段 1 的计算中使用的相同 ¹²。
4.2.3 TI 模拟
所有模拟均采用遵循 9 点高斯求积的 λ 进度表和软核势。每个 λ 窗口通过 2000 步最小化、随后在 NVT 系综中加热 50ps 以及在 NPT 系综中进行 300ps 的密度平衡来平衡。所有生产模拟均采用计算资源的动态优化13,以最小化模拟的计算成本。该方法利用短的初始模拟,随后通过 Jensen-Shannon 距离对耦合势导数时间序列的两个连续半部分进行迭代自动平衡检测29和收敛测试,如果未满足收敛标准,则进行额外的模拟。对于大多数模拟,初始模拟长度为 2.5ns,额外模拟长度为 0.5ns,Jensen-Shannon 收敛标准为 0.1。对于在最后一次 AL 迭代中进行的几个模拟,使用 1.0ns 的初始模拟长度和 0.25ns 的额外模拟长度以进一步加速模拟。当有额外资源可用时,进行各种转化的多个重复,优先考虑那些初始计算出的相对结合自由能为负的转化。当进行转化的多个重复时,通过集合方法计算 ΔΔG,其中给定 λ 窗口的每个梯度时间序列被单独平衡和去相关,然后组合所有值以确定整体梯度时间序列平均值。
4.3 AL 文库形成:ML 引导选择
基于机器学习模型的推荐,从 AL 集合中迭代采样用于 MD TI 模拟的分子,该模型以与我们之前的工作类似的方式利用主动学习方法14。在 AL 循环的每次迭代中,通过 AutoML 方法训练机器学习模型以预测绝对结合自由能。然后使用性能最高的模型筛选 AL 集合以寻找预测具有优异绝对结合自由能的分子。将 AL 前集合中分子的计算出的相对结合自由能转换为绝对结合自由能,并用于初始化 AL-RBFE 工作流程。
4.3.1 分子表示和 ML 算法
使用了以下特征化技术:1)RDKit 分子指纹(路径长度为 7 且二进制向量长度为 2048 的路径指纹,例如 RDKFP7_2048),使用 RDKit;2)摩根指纹(半径为 3 且二进制向量长度为 2048 的扩展连接性指纹,例如 ECFP6_2048),使用 RDKit;3)具有默认参数的 3D 分子指纹 E3FP30;4)使用 RDKit 的二进制向量长度为 1024 的药效团指纹(2D)(ph4fp2D_1024);5)使用 RDKit 的二进制向量长度为 1024 的药效团指纹(3D)(ph4fp3D_1024)。
使用了 scikit-learn 库31中实现的以下 ML 算法:1)线性回归;2)随机森林;3)具有 Tanimoto 核的高斯过程回归。使用内环 5 折交叉验证网格搜索进行最佳超参数选择。
4.3.2 机器学习建模
在 AL 循环的每次迭代中,使用所有具有可用绝对结合自由能(相对结合自由能转换为绝对结合自由能)的分子作为目标变量训练机器学习模型。在每次迭代中,基于分子表示和机器学习算法组合的留一法交叉验证(LOOCV),选择具有最高 R² 的机器学习模型(分子表示和算法的组合)。选择算法和分子表示后,在整个数据上重新拟合模型;然而,对于 AL 迭代 1-6,仅在命中分子 1 的衍生物上训练机器学习模型,并且选择的模型仅用于筛选 AL 集合的命中分子 1 的衍生物(n=16,101)。对于 AL 迭代 7,在命中分子 1 和 2 的衍生物上训练机器学习模型,并且选择的模型用于筛选整个 AL 集合。以贪婪方式进行分子选择,获取具有最负的 ML 预测绝对结合自由能的化合物。每个迭代的 AL 细节如表 S4 所示。
4.4 用于实验验证的分子选择
根据挑战预算(75 个分子或 10,000 美元,以先到者为准)选择用于实验验证的分子。仅基于计算出的最负的绝对结合自由能选择 70 个分子(67 个命中分子 1 的衍生物和 3 个命中分子 2 的衍生物),其余 5 个分子在具有负绝对结合自由能但偏向化学多样性的命中分子 2 的衍生物中选择。选定的 75 个分子向 Enamine 化学供应商询价。所有 75 个询价分子均通过初始供应商质量控制,并满足挑战预算。