
近日,Enamine 科学研究所在预印本平台 ChemRxiv 发表了题为《偕二氟取代饱和双环胺的物理化学性质 —— 药物发现中的高级砌块》的研究论文。该文系统阐述了这一系列化合物的相关性质,以下是对其核心内容的解读:https://doi.org/10.26434/chemrxiv-2025-bzd76
偕二氟取代饱和双环胺的物理化学性质 —— 药物发现中的高级砌块
摘要
本文探讨了偕二氟取代对最大环含 6-8 个原子的饱和双环胺物理化学性质的影响。氟化对质子化物种酸性(pKa)的影响与基于缺电子取代基诱导效应的直观预测相符。相比之下,偕二氟取代可能升高或降低亲脂性(LogP),其影响取决于双环核心的结构,并受 C-F 键与相邻 C-H 键空间取向的调控。
引言
饱和环胺被广泛认为是药物化学中的优势骨架,因其具有良好的药代动力学特征、代谢稳定性和构象刚性[1]。对此类骨架进行结构修饰是药物发现中的基本策略,通常旨在提高靶点亲和力、减少脱靶相互作用,或优化溶解度、亲脂性、膜通透性等物理化学参数。在这一背景下,两种互补策略占据主导地位:(1)通过环融合或桥连接实现结构刚性化(在本文中形成双环胺);(2)引入氟取代基(尤其是偕二氟亚甲基),以微调电子性质和分子行为[2-5]。
通过环融合实现构象锁定的策略已成功应用于多个临床候选药物和获批药物。例如,选择性 BCL-2 抑制剂 Sonrotoclax 利用双环7-氮杂螺[3.5]壬烷母核来调节关键药效团的最佳距离和空间方向[6];出于相同目的,2-氮杂螺[3.3]庚烷被用于设计靶向IRAK4 和 IMiD 底物的双功能降解剂KT-413[7];类似地,靶向认知功能障碍的11β-HSD1 抑制剂Xanamem(UE2343)采用桥连哌啶骨架,通过降低分子柔性和外排风险来改善中枢神经系统穿透性[8]。
与此同时,在过去几十年的农用化学品和药物开发中,将氟原子(特别是 CF₂单元)引入有机骨架已成为一种强大工具[9-13]。在众多应用中,含氟基团可调节宏观物理化学性质[14-19]、稳定所需结构[20,21] 或改善药物的药代动力学参数[22]。这些特性已在多种化合物中得到应用,如DPP-4抑制剂利格列汀(Gemigliptin)[23]、Kras家族调节剂 BBO-8520 [24]以及脑穿透性 ATM 抑制剂WSD0628[25]。
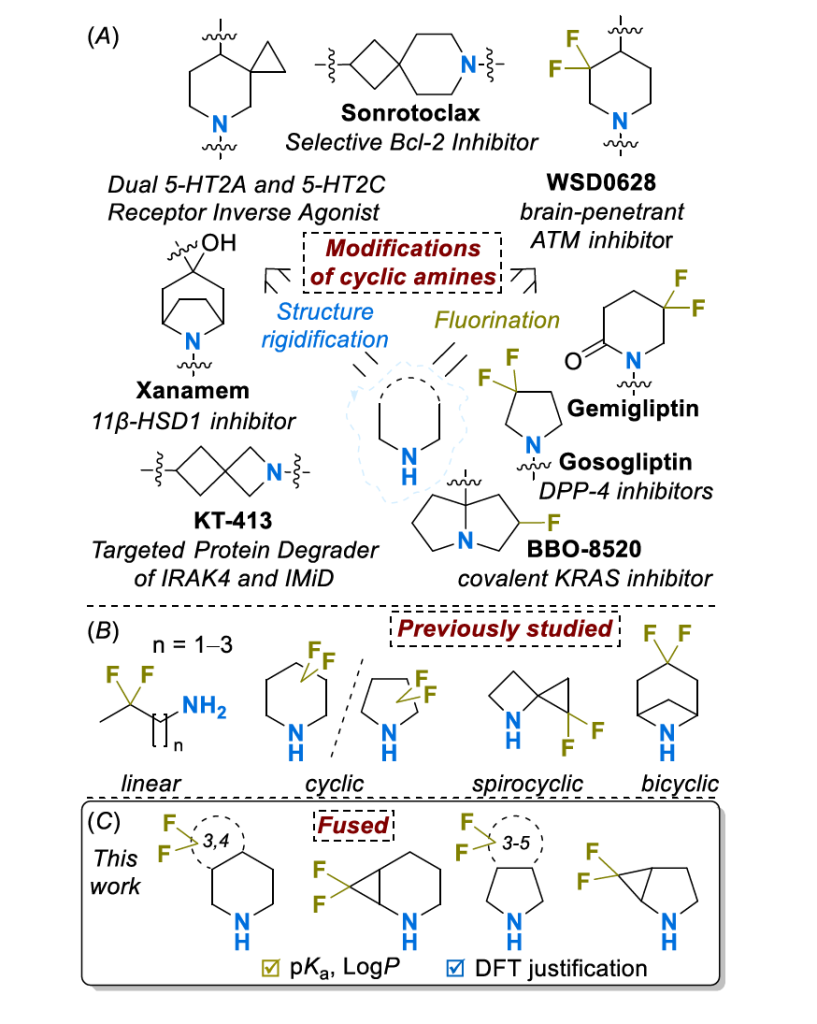
尽管双环胺和偕二氟基团各自的效用已得到证实,且与其他(非)环饱和胺与偕二氟亚甲基的组合不同 [26-29],但这两种策略的结合 —— 即含偕二氟片段的饱和双环胺 —— 尚未得到系统研究。本文考察了偕二氟取代对稠合饱和双环胺关键物理化学参数(即质子化物种的酸性(pKa)和模型酰胺衍生物的亲脂性(LogP))的影响。基于所得数据,揭示了氟对化合物亲脂性的复杂影响,为分子性质的精细调控提供了新方法。
结果与讨论
数据集:
本研究首先选取了一组可 “现成获取” 的偕二氟取代稠合双环胺(图 2 中黑色结构)。此外,还纳入了此前报道的桥连双环胺数据(图 2 中灰色结构)[27,30,31]。所得化合物集可根据分子中 C+N 原子总数分为三类 —— 哌啶类似物(1a 和 1b)、氮杂环庚烷类似物(1c-1f)以及氮杂环辛烷 / 氮杂环壬烷类似物(1g-1j)。这一参数对亲脂性分析尤为重要,因为它与提供正 LogP 增量的 CH/CH₂单元数量相关。
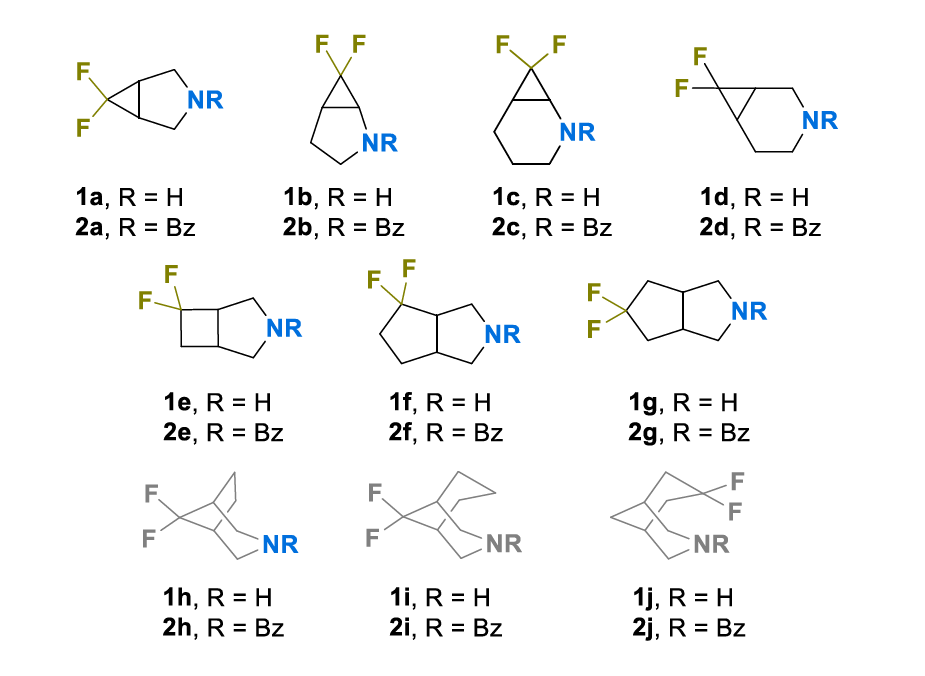
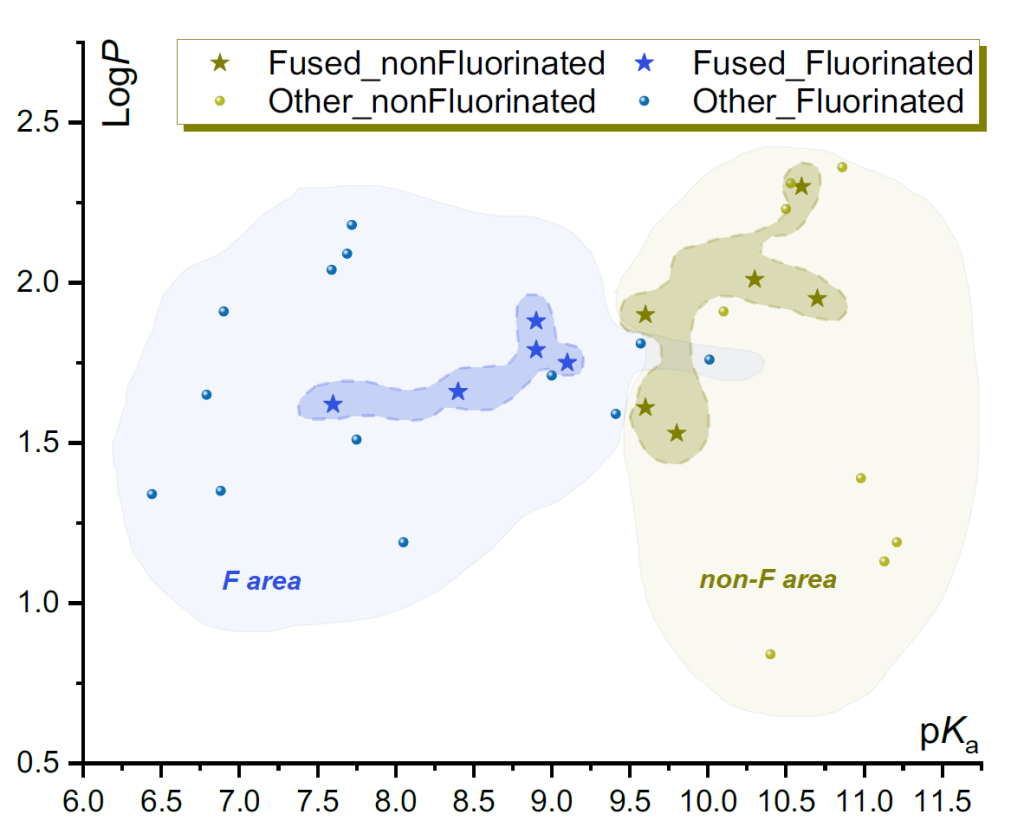
采用已建立的实验方案,对胺类化合物 1(以盐形式使用,通常为盐酸盐)进行了酸碱滴定,并测定了其模型苯甲酰胺衍生物 2 的正辛醇 / 水分配系数 [32]。所得 pKa 和 LogP 值汇总于表 1。为初步直观评估偕二氟取代稠合双环胺的物理化学特征,将所得数据与此前报道的线性伯胺 [28]、单环 [27]、桥连 [30,31] 或螺环 [26] 仲胺的数值一同绘制在 LogP-pKa 坐标中(图 3;所用化合物完整列表见支持信息中的图 S1)。根据 pKa 值,可清晰区分偕二氟取代化合物与非氟取代化合物。同时,所讨论的化合物(无论是否氟化)的亲脂性均在 1.5-2.2 LogP 单位范围内。
表 1. 化合物 1 和 2 在 21℃时的 pKa 值与 LogP 值。
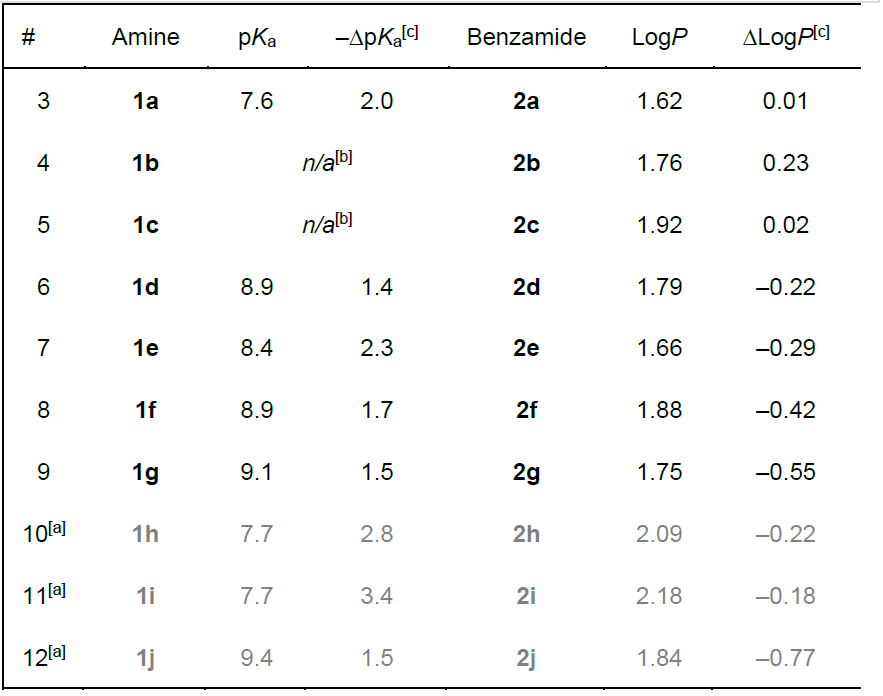
[b] 由于游离胺 1b 和 1c 的稳定性有限,未能获得其 pKa 值。
[c] 偕二氟化后观察到的 pKa 下降及 LogP 变化(与非氟化类似物相比)。
酸碱性质:所得偕二氟取代胺质子化物种的酸性值在 6.8 - 9.4 pKa 单位范围内(图 4)。遗憾的是,对于含偕二氟环丙烷部分且与氨基相邻的胺类 1b 和 1c,由于其游离碱稳定性有限,未能获得可靠的滴定结果。这一常见问题此前已有报道 [33],因此这些化合物未纳入分析。还计算了每种化合物相对于其非氟取代类似物的pKa 降低值(–ΔpKa)(非氟取代稠合双环胺的数据图 4. 质子化胺 1a–j 经偕二氟化后的 pKa 下降值(–ΔpKa)。由 Bohdan V. Vashchenko 博士及其同事提供,将图 4. 质子化胺 1a–j 经偕二氟化后的 pKa 下降值(–ΔpKa)。在其他地方详细讨论)。
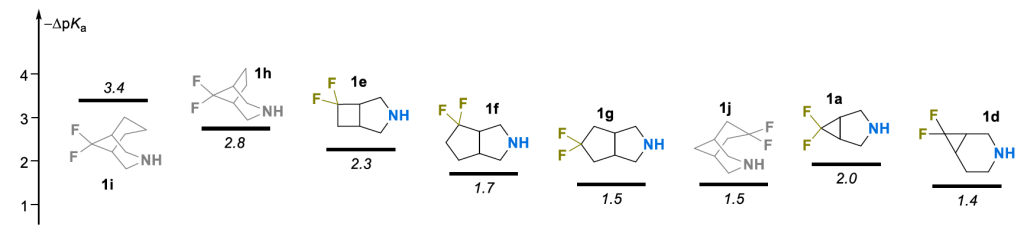
总体而言,观察到的 pKa 降低与氮原子和CF₂部分之间的最短通过键路径长度相关,这表明氟的诱导效应对化合物的酸碱性质起主导作用。事实上,通过引入额外的 CH₂单元(1e,1f,1g)增加一条或两条最短通过键距离,导致–ΔpKa 值稳步降低(分别为 2.3, 1.7,1.5)(图 4)。值得注意的是,分子形状相似的化合物 1g 和 1j 在偕二氟取代后 pKa 降低值相同(–ΔpKa=1.5)。
环丙烷衍生物 1a 和 1d 未遵循一般趋势:尽管两个含杂原子的片段位置相对接近,但其–ΔpKa 值(分别为 2.0 和 1.4)比同系物系列的预期值小约1个单位。这可能与环丙烷环众所周知的部分不饱和特性有关,因此共振效应可能也对这些双环分子的电子结构有贡献 [34]。然而,若单独考虑化合物 1a 和 1d,其 pKa 降低也符合上一段所述的一般逻辑。
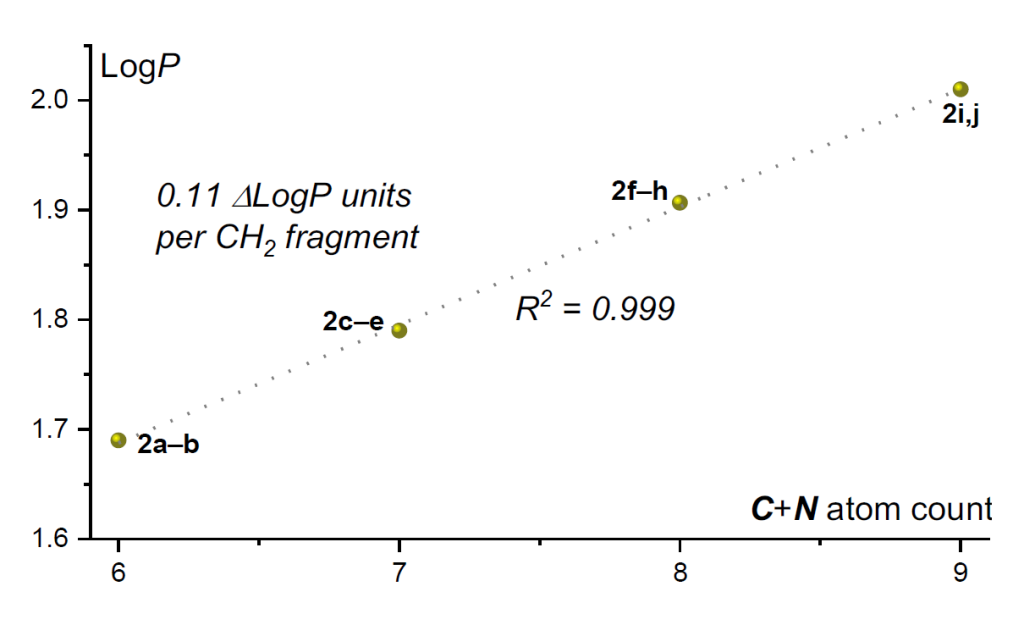
亲脂性:如上所述,偕二氟取代苯甲酰胺 2a-j 的LogP 值在 1.5-2.2 单位范围内。平均亲脂性与 C+N 原子总数呈极佳的线性相关性(图5,R²=0.999)。每增加一个碳原子,LogP 值增加 0.11 单位,这显著低于由 Hansch 亲脂性常数得出的 CH₂基团增量(约 0.5)[35]。
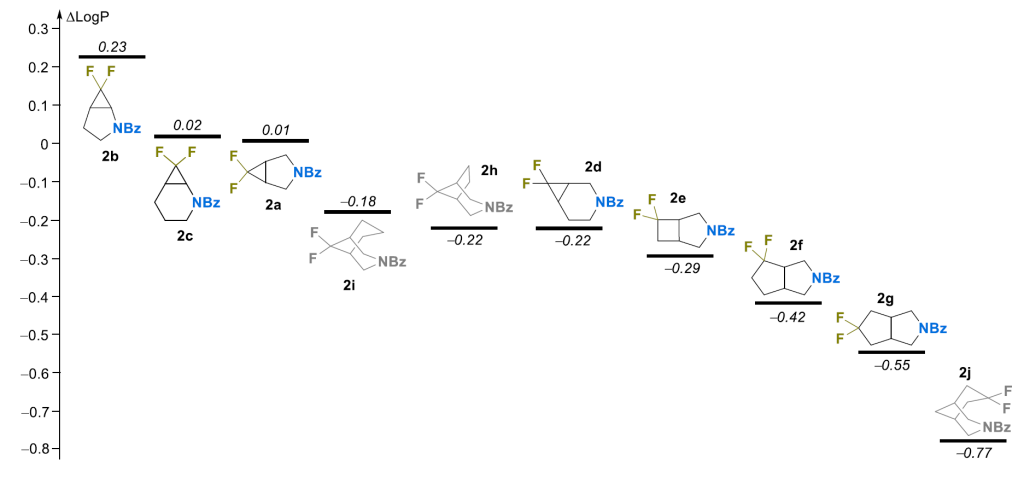
偕二氟取代后的 LogP 变化(ΔLogP)遵循复杂趋势(图 6)。该参数与氮原子和 CF₂部分之间的最短通过键距离以及包含这两个片段的环的大小均有良好相关性。因此,在 β 位的偕二氟取代导致亲脂性增加(平均 0.13 LogP 单位),而在 γ 和 δ 位的偕二氟取代则导致亲脂性降低。这一结果与各种同源偕二氟取代无环官能化衍生物的研究结果一致 [19,36],但与偕二氟环烷烃衍生的苯甲酰胺或苯胺不同 [28]。C+N 原子总数似乎也对 ΔLogP 值有影响,这在化合物 2i 和 2j 以及该系列的其他一些代表物中较为明显。
分子静电势表面(MEPS)分析:在许多先前的研究中,官能团偶极矩的相对取向被用于解释观察到的亲脂性趋势 [28,36-38]。显然,这种方法并非总能适用于化合物 2a-j。实际上,在该系列中,有许多例子表明更对称的化合物具有更低的 LogP 值(例如 2a 与 2b 或 2g 与 2f)。
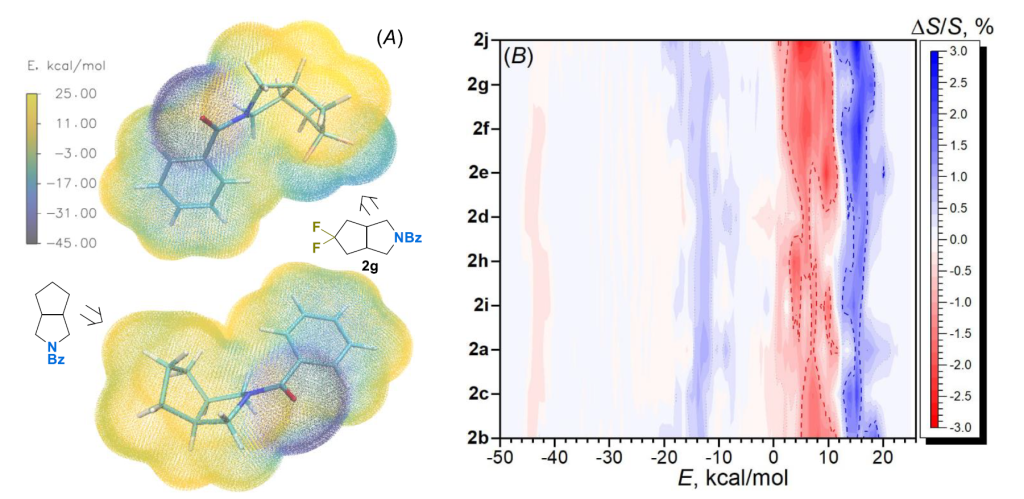

不久前,Hunter 及其同事应用分子静电势表面(MEPS)分析来评估溶剂化能,而溶剂化能又与亲脂性直接相关 [39,40]。最近,我们使用这种方法解释了氟化对环丁烷衍生物系列中 LogP 的影响 [41]。我们计算了化合物 2a-j 及其非氟取代类似物的静电势分布(图 7,A 为代表性例子2g 及其非氟取代类似物,支持信息中的图 S6-S22)。图 7,B 以热图形式展示了它们的差异,即偕二氟取代后观察到的相应 MEPS 面积的相对变化(ΔS/S)(另见支持信息中的图 S4)。因此,在所有情况下,引入氟原子均导致分子表面极化增强,同时伴随着两个不同区域的显著扩大 —— 具有较高正电势(10 - 20 kcal/mol)和负电势(–20 - –5 kcal/mol)的区域。同时,电势在–5 - 10 kcal/mol 范围内的 “中性” 表面积(在非氟取代化合物中定义明确)显著减少。通常还观察到极负区域(–45 - –40 kcal/mol)的 MEPS 面积减少。
MEPS 的可视化和高极化区域的定位为观察到的数据提供了可能的物理解释(图 7,C 为代表性例子2g)。实际上,在所有研究的化合物中,“高负”(–45 - –40 kcal/mol)和 “负”(–20 - –5 kcal/mol)MEPS 区域分别位于 C=O 和 C-F 片段,这显然与相应杂原子的强吸电子性质有关。相反,“高正” MEPS 区域(10-20 kcal/mol)的位置因化合物结构而异,但通常出现在靠近氟化部分的某些氢原子上(见支持信息中的图 S23-S32)。
表 2. 偕二氟化后观察到的实验性亲脂性变化(ΔLogP/LogP),及其与不同静电势范围内相应计算的相对 MEPS 面积变化(ΔS/S)的相关性。
对氟化引起的极性面积变化的定量分析揭示了化合物结构与 ΔLogP 之间关系的一些有趣见解。将亲脂性相对变化(ΔLogP/LogP)与特定电势值下的 MEPS 面积相对变化 ΔS/S(%)作图,发现在所研究的数据集中存在两个不同的亚组(表 2,支持信息中的图 S5)。因此,对于不含环丙烷环的双环化合物(2e-j),观察到 ΔLogP/LogP 与 “中性” MEPS 面积(E=–5 - 10 kcal/mol,对应弱极化 C-H 键)的减少呈显著正相关(R²=0.91)。还发现 ΔLogP/LogP 与高负区域(E=–45 - –40 kcal/mol,对应氧原子)的减少呈良好负相关(R²=0.94)。同时,ΔLogP/LogP 与 E=10 - 20 kcal/mol(对应高极化 C-H 键)和 E=–20 - –5 kcal/mol(对应氟原子)范围内的 MEPS 面积相对增加呈较不显著的负相关,R² 分别为 0.76 和 0.84。
所得结果证实,对于化合物 2e-j 系列,MEPS 极化程度越高,ΔLogP 负值越显著。偕二氟取代导致 C=O 键(其非氟取代类似物中几乎是唯一的强极性片段)极化降低,这似乎是影响化合物亲脂性的重要因素。实际上,在化合物 2e-j 中,CF₂与酰胺片段之间的距离(通过两条最短 C-C 键路径)越长,氟原子对 C=O 基团的吸电子诱导效应越弱,进而导致其极化程度越高,化合物亲脂性越低。当然,氢原子的正极化增强以及与氟原子相关的负表面极化也很重要,因为它们中的至少一个(或两者)本身就是观察到 ΔLogP 负值的主要原因。
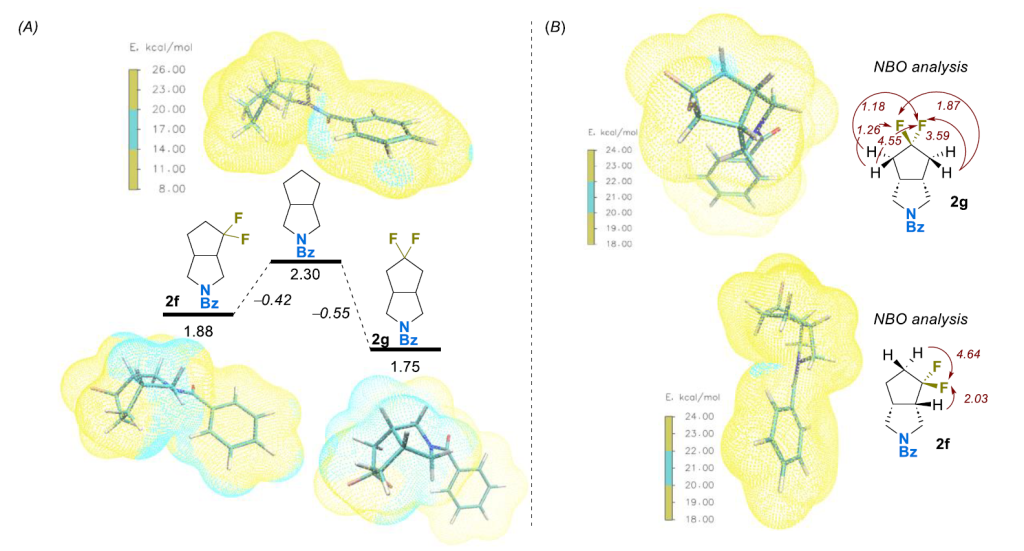
为进一步深入了解这些结果,我们比较了化合物2f、2g及其非氟取代类似物的 “高正” MEPS 区域位置(图8,A)。正如预期,亲脂性较低的2f和2g具有较大的带正电分子表面;与母体化合物相比,该区域主要位于靠近极性片段(偕二氟亚甲基和苯甲酰胺键)的氢原子上。上述化合物的自然键轨道(NBO)分析 [42] 显示,在偕二氟亚甲基相邻位置的C-H键存在显著的非路易斯离域能(E_NL=1 - 5 kcal/mol),表明可能通过 σC−H→σ*C−F超共轭使邻位 C-H 键极化(图 8,B)[19,43]。由于此类相互作用的效率强烈依赖于相应 C-H 和 C-F 键的相对取向,所研究化合物的双环性质因其构象限制而对亲脂性产生复杂的精细影响,这与我们的实验观察一致。因此,与2g相比,2f 中观察到更多的相互作用,这可能导致其LogP更低。
含环丙烷的化合物 2a-d 呈现出截然不同的趋势。与 2e-j 的情况相反,“中性” 分子表面积(E=5 - 10 kcal/mol)的相对变化与 ΔLogP/LogP 呈显著负相关(R²=0.94);同时,“高正” 区域(E=10 - 20 kcal/mol)的 ΔS/S 与亲脂性呈正相关(R²=0.92)。需要注意的是,尽管 R² 值较高,但由于数据集规模小(仅 4 种化合物),这些相关性需谨慎分析。此外,在 ΔLogP/LogP 与氧原子对应的 “高负” 区域(E=–45 - –40 kcal/mol)或氟原子对应的 “负” 区域(E=–20 - –10 kcal/mol)的 MEPS 面积变化之间未观察到相关性。
这些数据表明,氟原子引起的表面极化不一定会增加分子在水相中的溶剂化(从而降低 LogP)[44]。文献中提出了若干原因,包括氟原子极低的极化率降低氢键形成倾向和其他非共价相互作用 [22]、溶剂空腔形成成本与氢键形成收益之间的平衡[44],以及氟原子比氢原子稍大从而增加分子总疏水表面积[44,45]。显然,在偕二氟环丙烷的情况下,环丙烷片段的作用(已知其会降低亲脂性[46])对化合物的LogP和偕二氟取代后观察到的净ΔLogP值至关重要。后一种效应可能与氟原子因与环丙烷环的稳定相互作用而极化率更低有关[47],或者与小而相对缺电子的环丙烷部分周围溶剂 “壳层” 的破坏有关 [46]。综合来看,这些效应可能阻碍了与水分子的有效相互作用,从而增加了化合物的疏水性。另一方面,在本研究中其他化合物中大量观察到的可能降低LogP的效应(例如邻位C-H键极化)在这种情况下似乎不足,被环丙烷环的影响所掩盖。结合我们此前关于α-氟取代饱和碳环 / 杂环苯胺[48]和饱和含氧杂环[49]亲脂性的研究结果,我们可以推测上述观察可能属于更普遍的趋势:在饱和分子中靠近极性基团的位置引入氟原子以增加亲水性,实际上可能降低化合物的LogP。
结论
本文详细评估了偕二氟取代对饱和双环胺物理化学性质的影响。对酸性趋势的系统分析表明,酸碱性质与CF₂和 NH₂⁺官能团之间的 C-C 键数量呈良好相关性,这可通过吸电子偕二氟亚甲基片段的诱导效应合理解释(尽管双环偕二氟环丙烷衍生物与一般趋势不太吻合)。一系列模型苯甲酰胺的亲脂性与重原子(C+N)数量相关,但偕二氟取代对LogP的影响复杂。因此,平均ΔLogP 值根据CF₂与酰胺部分之间的距离呈现相反的符号 ——β,β- 二氟取代衍生物亲脂性略有增加,而其 γ,γ- 和 δ,δ- 二氟取代类似物亲脂性较低。
进一步的分子静电势表面(MEPS)分析表明,偕二氟取代增加了分子表面的整体极化,特别是某些氢原子上的 “高正” 区域,这与大多数所研究双环化合物的ΔLogP 趋势相关。研究表明,σC−H→σ*C−F超共轭可能对观察到的 MEPS 变化很重要,这在具有高度构象限制的双环系统中尤为显著。同样,双环偕二氟环丙烷衍生物呈现出不同的趋势;在这种情况下,通常观察到亲脂性增加,且与MEPS分析数据不相关(甚至呈反相关)。
这些结果表明,分子表面极化(与溶剂化能相关)不应被视为决定所研究系列中偕二氟取代对化合物LogP影响的唯一因素。我们认为,结合上述文献先例,我们的结果展示了氟调节有机分子亲脂性的双向能力[44]。特别是,由于氟原子极低的极化率和比氢原子稍大的尺寸,偕二氟亚甲基可能增加亲脂性;相反,通过诱导效应和 / 或超共轭使邻位基团极化应导致LogP降低。在饱和双环系统中,由于构象限制,只有当相应 C-H和C-F键取向有利时,这些相互作用才会显著表现出来。
我们希望所得结果能为偕二氟取代饱和双环胺作为药物发现项目中的高级砌块的合理应用提供帮助,并有助于理解饱和有机氟化合物的物理化学性质。
支持信息
作者在支持信息中引用了额外的参考文献 [32,42,50-62]。
参考文献(略)