导读:本文以氮杂螺环替换布比卡因哌啶母核,合成3种衍生物。动物试验证实衍生物保留局麻活性、心脏毒性显著下降,因药物更少富集心脑组织。依托Enamine开发的螺环氮杂环丁烷特色分子砌块完成骨架改造,证实该砌块可作为哌啶优良生物电子等排体,为药物分子改造提供思路。
1、摘要
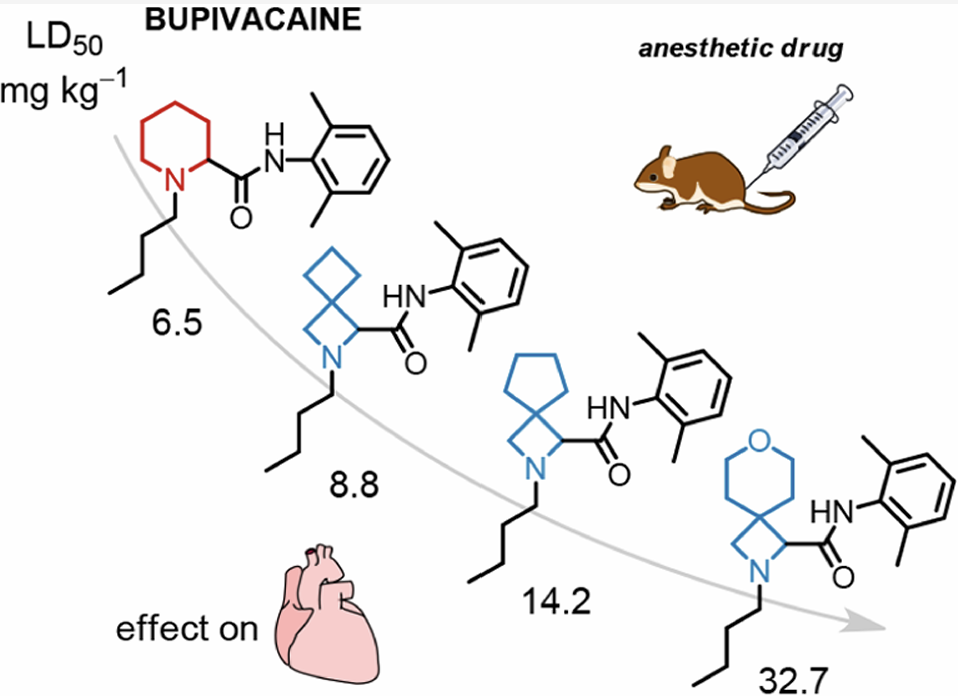
局麻药布比卡因入血后可引发全身性毒性,诱发心脏不良反应,是临床亟待解决的难题。本研究将布比卡因分子中心的哌啶环替换为线型氮杂螺环骨架,合成系列衍生物;小鼠体内药效试验证实衍生物保留合格的局部麻醉活性。三类螺环改造产物毒性显著下降,半数致死剂量(LD₅₀)分别提升至原药的1.3倍、2.2倍、5.0倍。离体豚鼠心脏实验中衍生物QRS波增宽程度更轻微,证实毒性降低源于心脏毒性减弱。小鼠药代数据显示:螺环衍生物与血浆结合亲和力更高,而布比卡因更易富集于脑与心脏组织。综上,螺环改造策略适用于局麻药结构优化,螺环氮杂环丁烷可作为哌啶环的优选生物电子等排体用于药物结构修饰,值得深入开发各类螺环型布比卡因衍生物。
2、前言
局部麻醉药(局麻药)通过多重作用机制抑制神经末梢兴奋、阻断外周神经冲动传导。外周神经元依靠电压门控钠通道产生动作电位以传导痛觉:神经受刺激时钠通道开放、胞外钠离子内流,启动电信号;局麻药结合静息态钠通道,阻断动作电位传导、实现镇痛。局麻药临床应用广泛,多用于门诊手术区域阻滞、牙科麻醉、急慢性疼痛管控;除此以外,该类药物还兼具抗肿瘤、抗炎、抗菌、抗心律失常等药理活性。
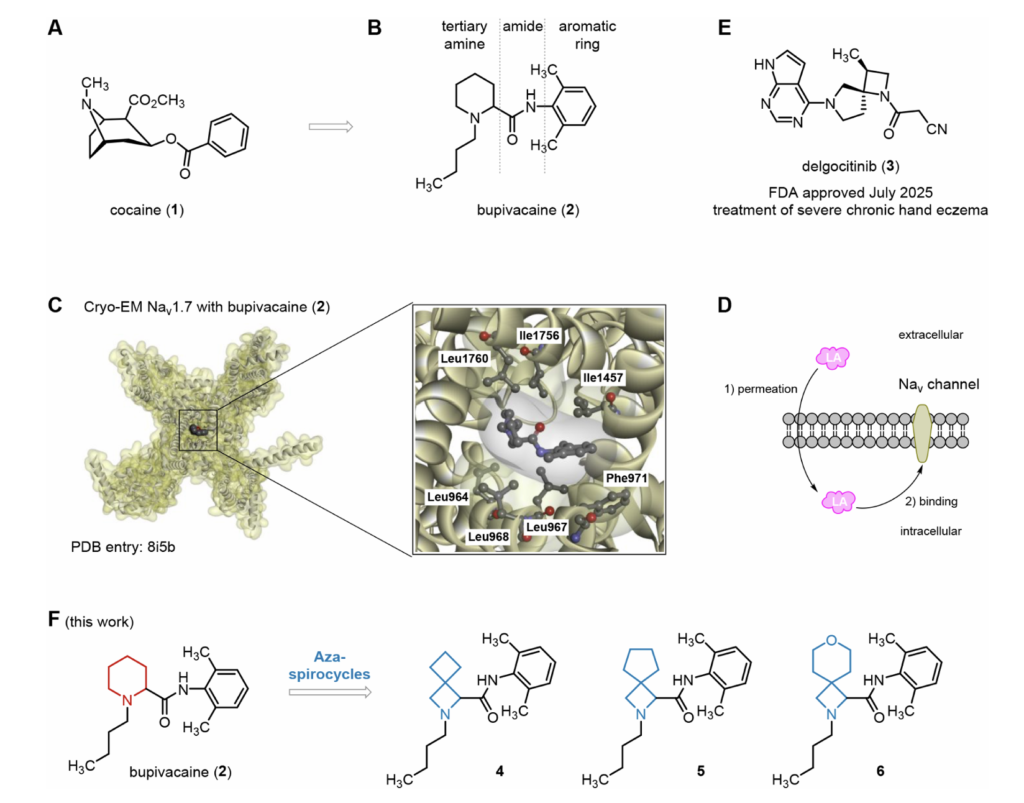
现代合成局麻药源自可卡因,分子结构固定包含三部分:叔胺结构、酯键 / 酰胺连接片段、疏水性芳香环。布比卡因是酰胺类经典局麻药,1957年首次合成,分子仅含21个非氢原子,至今仍是临床主流局麻药物。冷冻电镜结构显示:布比卡因从胞内侧结合人源Nav1.7钠通道,堵塞通道胞内侧门控区并使通道收缩约1埃;该结合模式决定药物需要穿透神经细胞膜才能起效,同时布比卡因也是现有临床局麻药中脂溶性最高的品种。美国FDA在2011年批准多层囊泡脂质体布比卡因上市。
布比卡因最凶险的不良反应为心脏毒性,可诱发心脏传导阻滞甚至心搏骤停。其心脏毒性与药物非特异性结合心肌钾通道、钙通道相关,干扰心肌动作电位。因此,通过化学结构修饰降低布比卡因心脏副作用具备重要临床价值。
本课题组及其他研究团队长期开发螺环砌块,用于替换药物分子中的哌啶母核。螺环结构能够固定分子优势构象,同步优化水溶性、脂溶性,降低体内酶解速率。2025年7月FDA获批用于重度慢性手部湿疹的德拉戈替尼(Delgocitinib),就是螺环氮杂环丁烷骨架药物的成功范例。本工作设计3种螺环修饰型布比卡因衍生物(化合物4~6),筛选得到心脏毒性更低、药代动力学优良的候选化合物。
3、结果与分析
合成化学
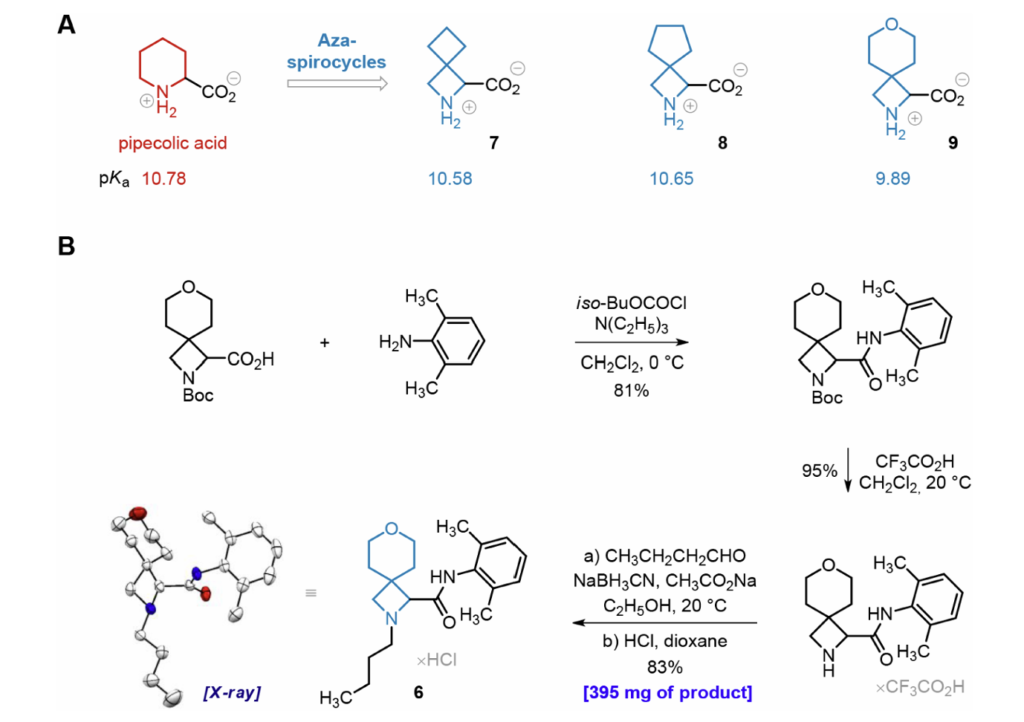
布比卡因母核依托哌可酸搭建:哌可酸氮端烷基化、碳端酰胺化得到目标结构。本课题前期已完成3种螺环哌可酸类似物(7~9)合成;酸碱测定表明:类似物7、9铵根pKa与哌可酸相差极小,含氧螺环9因环内氧原子场效应,pKa下降0.9个单位。
初期小量合成采用N-Boc保护氨基酸制备衍生物4~6,收率偏低、仅能得到毫克级产物;为满足动物大样本体内试验,优化全合成路线。以化合物6为例:第一步酰胺化收率81%;N-Boc脱保护收率95%;摒弃原先低收率碘丁烷烷基化,改用丁醛还原胺化,成盐酸盐后总三步综合收率64%,一次性制备395mg产物,且获得可用于单晶衍射的结晶样品。衍生物4、5采用相同工艺,分别制备1.33g、512mg。所有产物均为盐酸盐消旋体;上市布比卡因同样为消旋体,其中S-左旋布比卡因(左布比卡因)镇痛活性略优、毒性更低,R-右旋体心脏毒性更强,原因是右旋体与心肌钠、钾通道结合更快、亲和力更高。但临床试验证实消旋布比卡因整体药效与左布比卡因无显著差异,临床仍以消旋体为主,因此本研究体内药效与毒性试验均采用消旋衍生物,体外拆分单一异构体补充测试。
晶体结构解析
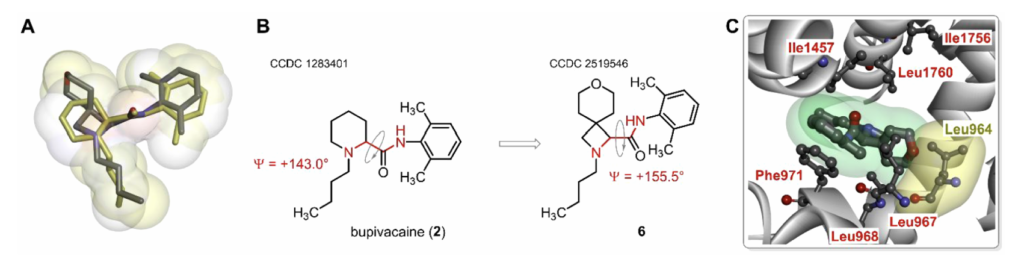
解析化合物6单晶衍射数据,并与已发表布比卡因晶体结构叠合对比:两者酰胺键空间取向高度重合;最大结构差异来自四氢吡喃含氧螺环的空间位阻,该片段伸入Nav1.7通道空腔,靠近高度保守的门控亮氨酸Leu964残基,存在空间位阻排斥;衍生物4、5的螺环体积更小,与通道空腔适配性更好。整体结构分析支持3种螺环衍生物作为布比卡因仿生衍生物继续开发。
理化性质
测试衍生物溶解度、脂水分配系数(pH7.4 下 logD)、血浆蛋白结合率。布比卡因叔胺pKa=8.2,生理环境下主要以质子化阳离子存在,但仍具备较强疏水性,实测pH7.4条件logD=2.7。脂溶性排序:5≥4>6>布比卡因;螺环母核普遍提升脂溶性,但6的环内氧原子显著降低亲脂性。脂溶性越高水溶性越差,化合物5脂溶性最高、水溶性最低,同时血浆蛋白结合率最高;高血浆蛋白结合通常延长局麻药神经阻滞时长,与药效正相关,本试验中全部衍生物血浆蛋白结合率均优于原型药布比卡因。
体外生化评价
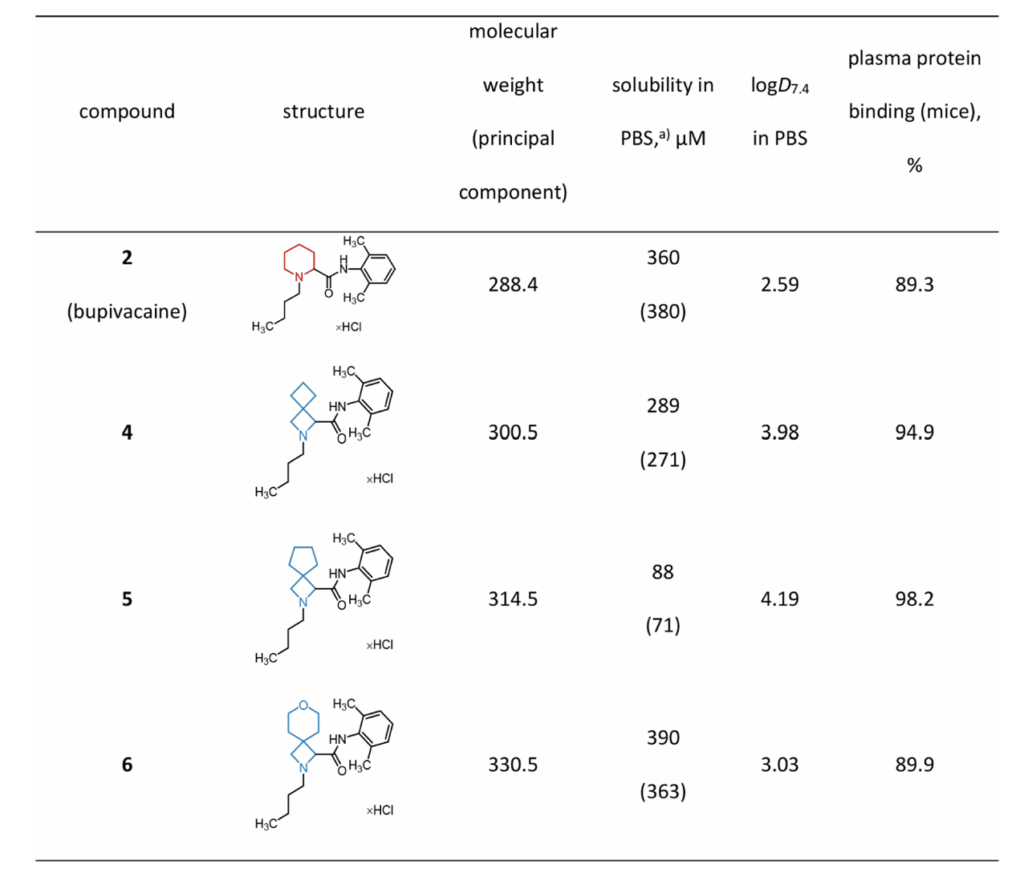
hERG通道抑制是药物心脏毒性关键筛查指标,化合物抑制hERG极易诱发QT间期延长、临床撤药。采用过表达hERG的HEK293细胞铊离子通量试验:25μM给药浓度下,所有螺环衍生物对hERG抑制能力均低于左布比卡因,且左旋异构体抑制活性普遍弱于右旋异构体,预示衍生物心脏毒性更低。
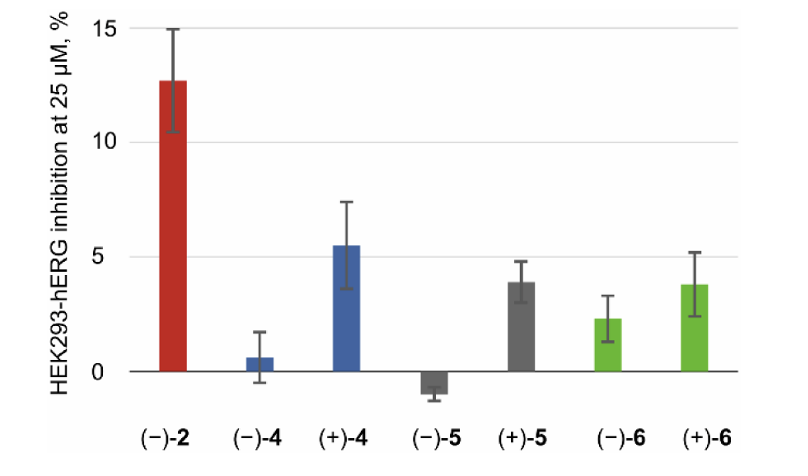
人肝微粒体稳定性试验:3种衍生物体内代谢速率均快于布比卡因,脂溶性最强的5代谢最不稳定。
MDR1-MDCKII细胞渗透性试验(模拟血脑屏障穿透能力):4种药物双向通透系数均为优良水平,外排系数接近1,无明显主动转运,均可穿透神经细胞膜发挥局麻作用。
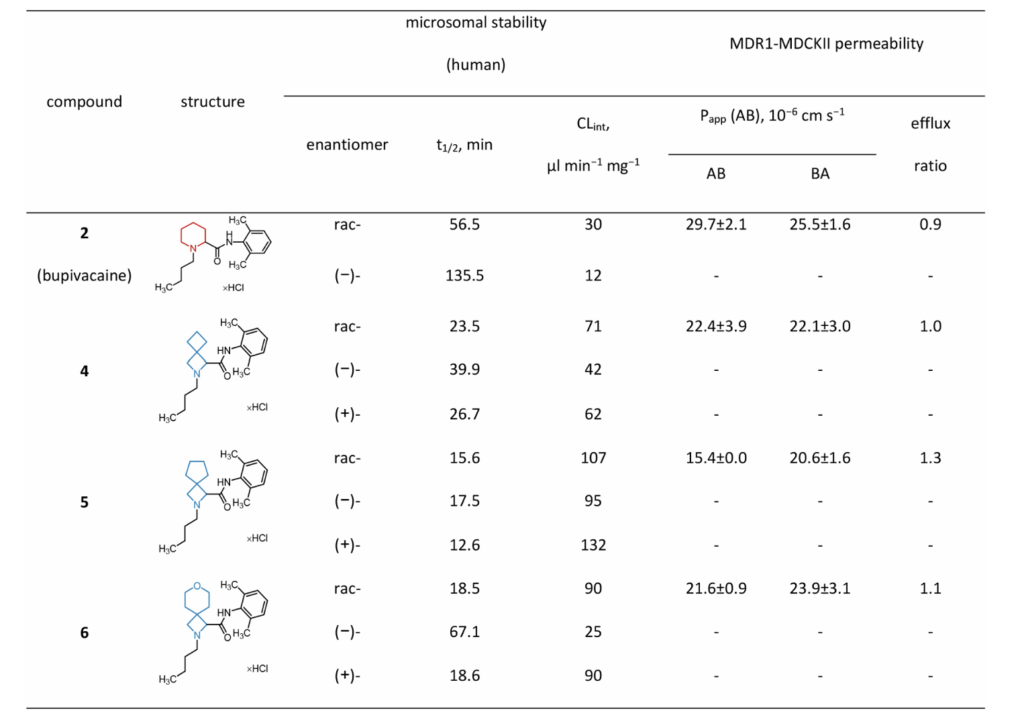
小鼠体内镇痛活性(甩尾热痛模型)
小鼠尾根部皮下给药,采用红外热辐射甩尾试验评价局部镇痛;因5水溶性差,给药配方添加20%聚乙二醇蓖麻油(Kolliphor EL)助溶,其余样品用pH6.1酸性生理盐水配制,给药剂量0.5mg/只、0.25mg/只。
0.5mg剂量:布比卡因药效在5~45min达峰值,90min回落至基线;衍生物4时效曲线与原型基本一致;6起效迅猛但药效15min后快速衰减;添加助溶剂的5镇痛时长、整体药效与布比卡因持平。基于药时曲线下面积(AUC)综合药效排序:4≈布比卡因≈5>6。
低剂量0.25mg组:布比卡因、4、5起效5~10min达最大镇痛,但药效衰减速度相较高剂量更快;特例为化合物6:高低剂量镇痛峰值无差别、整体作用短促。
临床常规联用肾上腺素延长局麻时效,因此开展布比卡因、6+5μg肾上腺素配伍试验:联用肾上腺素后6镇痛持续时间显著延长,整体药效与单用布比卡因相当,证实6本身具备合格局麻活性,仅单用时效不足。
尾静脉注射后小鼠热板试验:全部化合物无全身镇痛效果,仅局限于局部起效,符合局麻药成药特征。
急性整体毒性(LD₅₀)
小鼠尾静脉梯度剂量给药测定半数致死量:5mg/kg剂量下全部受试小鼠存活;布比卡因LD₅₀最低,毒性最高;衍生物4、5、6的LD₅₀分别为原型1.3倍、2.2倍、5.0倍,6整体毒性降幅最大。
亚致死剂量给药后连续14d饲养观察:短期给药后小鼠出现一过性惊厥(局麻药经典神经中毒表现),后续存活小鼠进食、体重、脏器系数与空白对照组无统计学差异;解剖心、肝、脾、肾等脏器无病理性脏器增重/萎缩。
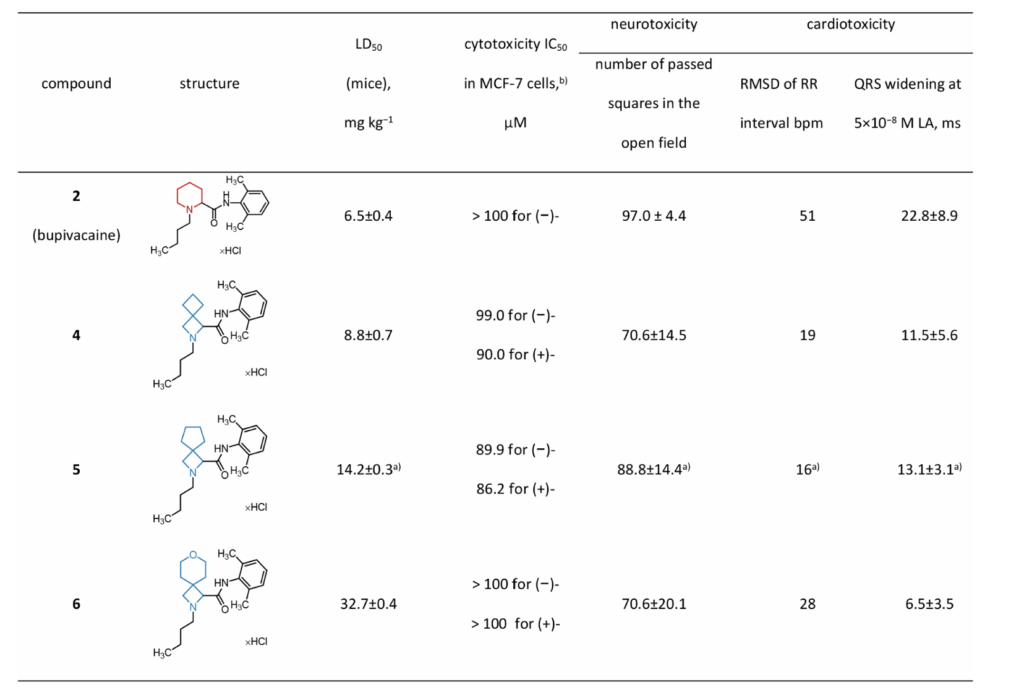
人乳腺癌MCF-7细胞体外细胞毒试验:细胞半数抑制浓度与动物体内急性毒性无相关性,提示体内毒性来源于心脏、神经等靶器官生理紊乱,而非直接细胞杀伤。
神经毒性评价
离体心脏毒性(朗根多夫灌流豚鼠心脏)
离体灌流豚鼠心脏模型,梯度给药监测心电图RR间期、QRS波宽度;QRS增宽是心肌传导阻滞、药物心脏毒性核心标志。
布比卡因可明显扰乱心率、大幅加宽QRS波(最高给药浓度下QRS增幅23%);衍生物引发的心电图异常幅度显著降低,QRS增宽程度排序:布比卡因>5≈4>6,与体内LD₅₀毒性排序完全吻合。证明衍生物全身毒性下降的核心原因是心脏毒性降低。
体内药代动力学
小鼠静脉等摩尔混药注射,分时段检测血浆、脑组织、心脏组织药物浓度:布比卡因在血浆清除更快,但在脑、心脏富集浓度远高于螺环衍生物;皮下给药结果趋势一致:布比卡因血浆暴露量最低,心、脑峰值浓度(Cmax)、药时曲线面积(AUC)显著偏高;衍生物4、5脂溶性更高但在心脑靶器官峰值浓度更低,更多滞留于血浆;6理化居中,组织分布介于两者之间。
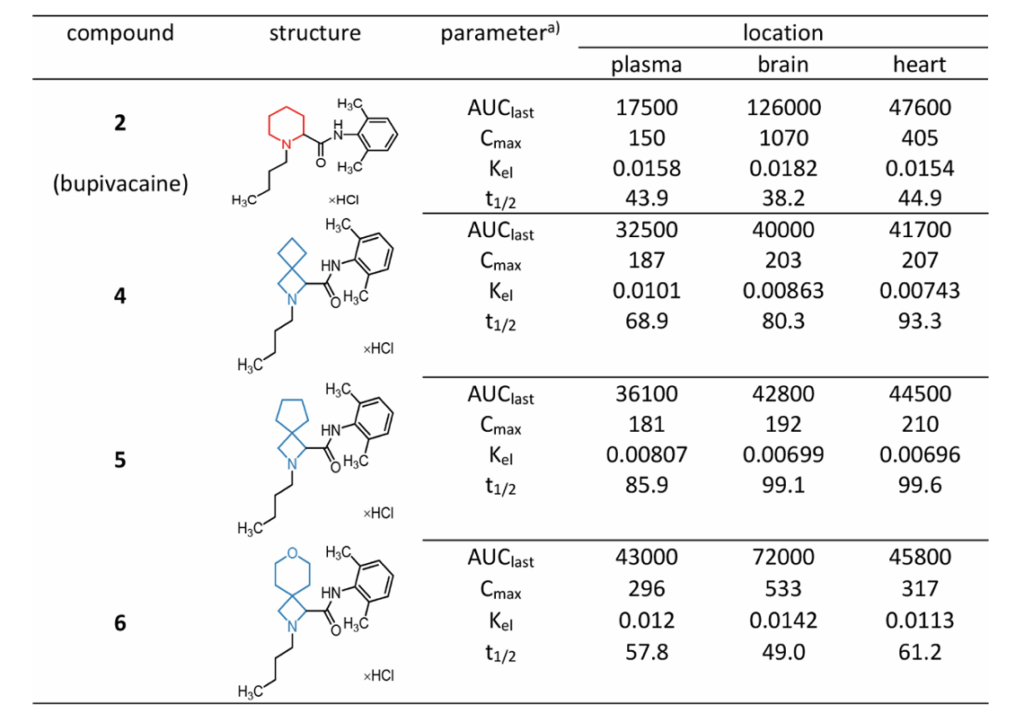
结论:布比卡因对心脏、脑组织高亲和力是其高心脏毒性的关键药代诱因;螺环改造改变组织靶向分布,减少心脑药物蓄积、降低毒副作用。
4、讨论
自19世纪末合成局麻药问世,局部麻醉广泛用于临床,但局麻药全身中毒(LAST)以心脏、中枢神经毒性为致命表现。布比卡因自上世纪60年代临床应用,儿童、孕产妇使用时中毒风险突出,由此催生罗哌卡因(布比卡因手性改造药物,90年代上市);但罗哌卡因仍无法完全规避心脏毒性风险。
近十年螺环骨架成为药物化学热门改造砌块:三维刚性结构固定优势构象,同步优化理化、药代属性;螺环氮杂环丁烷已大量用于临床在研新药(德拉戈替尼、JDQ-443等),大量文献证实螺环修饰可显著降低候选药物脱靶毒性、优化心脏安全性。
本研究以三类螺环替换布比卡因哌啶环,产物脂溶性整体上升、水溶性下降,高疏水的5需添加助溶剂给药;衍生物肝微粒体代谢稳定性弱于原型,但全部保留局部镇痛活性,仅6单用作用时活性减弱。药代数据显示螺环修饰后药物从心、脑向血浆重新分布,消除布比卡因皮下给药后组织药物骤升现象。
化合物4、5镇痛活性与布比卡因持平,毒性分别下降至1/1.3、1/2.2;6毒性降至原型1/5但单用药效偏弱:原因一是环内氧导致pKa下降至7.4,生理pH下活性质子化药物占比减少;二是螺环体积与Nav1.7通道门控残基产生空间排斥,削弱靶点结合。离体心脏QRS数据、hERG抑制结果共同证实衍生物心脏毒性全面改善,动物体内毒性降低与心脏安全性提升直接相关。
本研究证实哌啶→氮杂螺环是优化局麻药治疗指数的有效通用改造策略,为现有老药结构优化、低毒新型局麻药开发提供新思路。
5、结论
将布比卡因母核哌啶环替换为三种线型氮杂螺环,得到衍生物4~6:在保留局部镇痛活性前提下,整体毒性显著下降。体外hERG抑制减弱、离体心脏QRS波增宽程度降低,明确毒性改善源于心脏毒性下降;神经毒性无明显差异。药代研究表明:螺环衍生物在心、脑组织亲和力更低,减少高危脏器药物富集是布比卡因原型毒性偏高的重要诱因。
衍生物4、5毒性分别降至原型1/1.3、1/2.2且镇痛等效,皮下给药无组织药物暴发性峰浓度,药代特征更优。综上,饱和氮杂环替换为氮杂螺环是提升药物治疗选择性、降低脱靶毒性的高效药物设计手段。